Virtual Screening Pipeline Engine — Design Document¶
Overview¶
The Virtual Screening Pipeline Engine is the core workflow orchestration system of the ChemLib Platform. It allows users to define, configure, and execute multi-stage compound screening pipelines using a visual DAG (Directed Acyclic Graph) editor. Compounds flow through progressively expensive computational filters, from simple property checks (milliseconds) to molecular docking (minutes), creating a funnel that identifies the most promising drug candidates.
Concept: The Drug Discovery Funnel¶
Click diagram to zoom and pan:
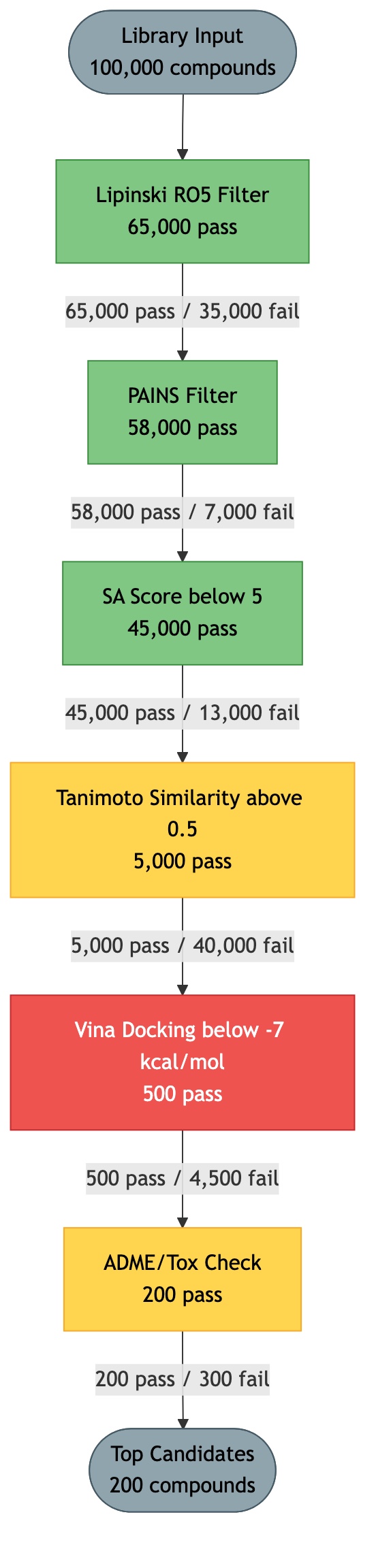
Drug discovery screening follows a funnel pattern. You start with a large compound library and progressively filter through increasingly expensive and selective computations. Each stage eliminates compounds that fail the criteria, reducing the set before more costly evaluations.
┌─────────────────────────────────────┐
│ Full Library (10,000 cpds) │ Property filters: ~1 ms/compound
└──────────────────┬──────────────────┘
▼
┌─────────────────────────────────────┐
│ Lipinski + PAINS + QED (7,000) │ ~3 ms total per compound
└──────────────────┬──────────────────┘
▼
┌─────────────────────────────────────┐
│ Similarity/Substructure (2,000) │ ~5 ms/compound
└──────────────────┬──────────────────┘
▼
┌─────────────────────────────────────┐
│ Pharmacophore/Shape Screen (800) │ ~1 s/compound
└──────────────────┬──────────────────┘
▼
┌─────────────────────────────────────┐
│ Molecular Docking (200) │ ~30 s - 5 min/compound
└──────────────────┬──────────────────┘
▼
┌─────────────────────────────────────┐
│ Rescoring / MM-GBSA (50) │ ~5 min/compound
└──────────────────┬──────────────────┘
▼
┌─────────────────────────────────────┐
│ ADME/Tox Prediction (30) │ ~1 s/compound
└──────────────────┬──────────────────┘
▼
┌─────────────────────────────────────┐
│ Visual Inspection (10-20) │ Human review
└─────────────────────────────────────┘
Key Principle: Cheap filters first, expensive filters last. This minimizes total computation time by eliminating as many compounds as possible before reaching the expensive stages.
Pipeline as a DAG (Directed Acyclic Graph)¶
Pipelines are defined as DAGs where each node is a filter/scoring step and edges define the flow of compounds between steps. This is more flexible than a simple linear pipeline because it supports:
- Branching: One filter's output feeds into multiple downstream filters
- Merging: Multiple filters' outputs combine before a downstream step
- Parallel paths: Independent filters can run concurrently
- Conditional routing: "pass" vs "fail" edges route compounds differently
Pipeline Configuration Model (Pydantic)¶
# chemlib/schemas/pipeline.py
from __future__ import annotations
from datetime import datetime
from typing import Optional, Any
from pydantic import BaseModel, ConfigDict, Field
class PipelineNodePosition(BaseModel):
"""Position of a node in the visual editor."""
x: float
y: float
class PipelineNode(BaseModel):
"""A single processing step in the pipeline."""
id: str # Unique node ID within the pipeline (e.g., "node_1")
type: str # "source" | "filter" | "score" | "dock" | "external" | "sink"
plugin_id: str # References a registered FilterPlugin name (e.g., "lipinski_filter")
label: str # Display label (e.g., "Lipinski RO5")
config: dict[str, Any] = {} # Plugin-specific parameters (threshold, reference_smiles, etc.)
position: PipelineNodePosition = PipelineNodePosition(x=0, y=0)
class PipelineEdge(BaseModel):
"""A directed connection between two nodes."""
source_node_id: str # ID of the source node
target_node_id: str # ID of the target node
condition: str = "pass" # "pass" | "fail" | "all"
# "pass": only compounds that passed the source node flow to target
# "fail": only compounds that failed flow to target
# "all": all compounds flow regardless of pass/fail
class PipelineDefinition(BaseModel):
"""Complete pipeline configuration."""
name: str
description: str = ""
target_id: int | None = None # Optional protein target (for docking nodes)
nodes: list[PipelineNode]
edges: list[PipelineEdge]
def validate_dag(self) -> bool:
"""Verify the pipeline is a valid DAG (no cycles)."""
# Topological sort; raises if cycle detected
...
def get_execution_order(self) -> list[str]:
"""Return node IDs in topological order."""
...
def get_source_nodes(self) -> list[str]:
"""Nodes with no incoming edges (pipeline entry points)."""
...
def get_sink_nodes(self) -> list[str]:
"""Nodes with no outgoing edges (pipeline exit points)."""
...
# --- Pipeline CRUD schemas ---
class PipelineCreate(BaseModel):
definition: PipelineDefinition
class PipelineResponse(BaseModel):
id: int
name: str
description: str
target_id: int | None
definition: dict # Full PipelineDefinition as JSON
created_at: datetime
updated_at: datetime
model_config = ConfigDict(from_attributes=True)
class PipelineListResponse(BaseModel):
items: list[PipelineResponse]
total: int
# --- Pipeline Run schemas ---
class PipelineRunCreate(BaseModel):
"""Start a pipeline run."""
compound_ids: list[int] | None = None # Specific compounds to screen
assembled_molecule_ids: list[int] | None = None # Specific assembled molecules
use_all_compounds: bool = False # Screen entire library
use_all_assembled: bool = False
class PipelineRunResponse(BaseModel):
id: int
pipeline_id: int
status: str # "pending" | "running" | "completed" | "failed" | "cancelled"
total_compounds: int
processed_compounds: int
current_node: str | None # Which node is currently being processed
started_at: datetime | None
completed_at: datetime | None
summary: dict | None # {"node_1": {"passed": 800, "failed": 200}, ...}
error_message: str | None
model_config = ConfigDict(from_attributes=True)
class PipelineRunResultResponse(BaseModel):
id: int
run_id: int
compound_id: int | None
assembled_molecule_id: int | None
node_id: str
passed: bool
score: float | None
metadata: dict | None # Arbitrary data (docking poses, interaction details, etc.)
created_at: datetime
model_config = ConfigDict(from_attributes=True)
class PipelineRunResultFilter(BaseModel):
node_id: str | None = None
passed: bool | None = None
min_score: float | None = None
max_score: float | None = None
sort_by: str = "score" # "score" | "compound_id" | "created_at"
sort_order: str = "asc" # "asc" | "desc"
limit: int = 100
offset: int = 0
DAG Example¶
A typical screening pipeline DAG:
[Source: Library]
│
▼ (all)
[Lipinski Filter]
│
┌───┴───┐
│ pass │ fail → (discard)
▼
[PAINS Filter]
│
┌───┴───┐
│ pass │ fail → (discard)
▼
[QED Score > 0.4]
│
┌───┴───┐
│ pass │ fail → (discard)
▼
[Tanimoto > 0.3 vs Reference]
│
┌───┴───┐
│ pass │ fail → (discard)
▼
[Vina Docking vs Pocket 1]
│
┌───┴───┐
│ pass │ fail → (discard)
▼
[PLIP Interaction Analysis]
│
▼
[Results Sink]
Filter Plugin Protocol¶
Click diagram to zoom and pan:
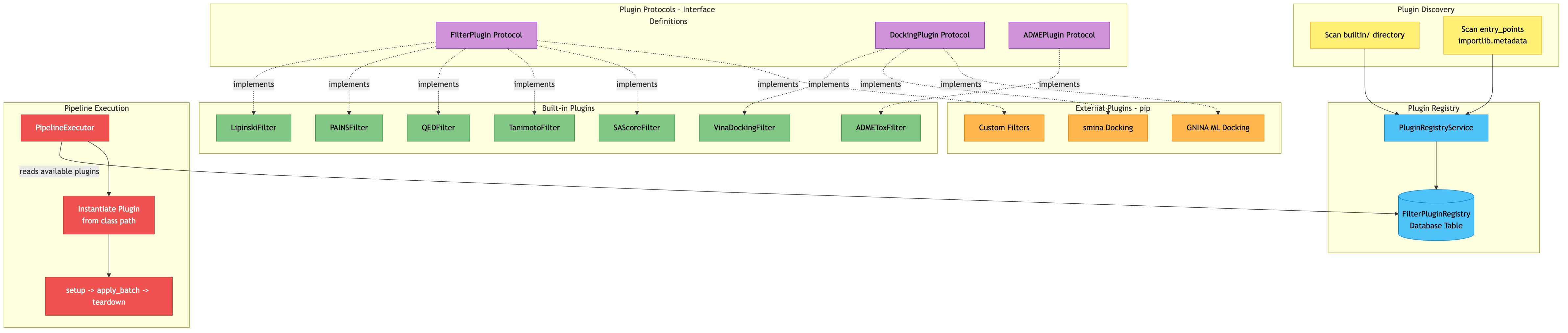
All filters implement the FilterPlugin protocol. This enables both built-in and third-party filters to integrate seamlessly.
chemlib/plugins/protocols.py¶
from __future__ import annotations
from typing import Protocol, runtime_checkable, Any
from dataclasses import dataclass, field
from rdkit import Chem
@dataclass
class PipelineContext:
"""
Shared context available to all filter plugins during a pipeline run.
Contains references to the protein target, binding site, and other shared state.
"""
run_id: int
pipeline_id: int
target_id: int | None = None
binding_site_id: int | None = None
binding_site_data: dict | None = None # Pre-loaded binding site info
receptor_pdbqt: str | None = None # Pre-prepared receptor for docking
reference_smiles: str | None = None # Reference compound for similarity
reference_fp: Any = None # Pre-computed reference fingerprint
extra: dict = field(default_factory=dict) # Plugin-specific shared state
@dataclass
class FilterResult:
"""Result of applying a filter to a single molecule."""
passed: bool
score: float | None = None
metadata: dict = field(default_factory=dict)
# metadata can contain:
# - docking: {"poses": [...], "best_score": -8.5, "interactions": {...}}
# - similarity: {"tanimoto": 0.72, "common_bits": 45}
# - property: {"value": 456.2, "threshold": 500.0}
# - adme: {"solubility": "high", "bbb_penetration": True}
error: str | None = None # Set if the filter encountered an error on this mol
@runtime_checkable
class FilterPlugin(Protocol):
"""Protocol that all filter plugins must implement."""
@property
def name(self) -> str:
"""Unique identifier for this filter (e.g., 'lipinski_filter')."""
...
@property
def display_name(self) -> str:
"""Human-readable name (e.g., 'Lipinski Rule of Five')."""
...
@property
def category(self) -> str:
"""Category: 'property', 'similarity', 'docking', 'adme', 'external'."""
...
@property
def description(self) -> str:
"""One-paragraph description of what this filter does."""
...
@property
def config_schema(self) -> dict:
"""
JSON Schema defining configurable parameters.
The UI will render a form from this schema.
Example:
{
"type": "object",
"properties": {
"allow_violations": {
"type": "integer", "minimum": 0, "maximum": 2,
"default": 0, "title": "Allowed Violations",
"description": "Number of Lipinski violations to allow"
}
}
}
"""
...
@property
def estimated_time_per_compound(self) -> str:
"""Estimated time: 'ms', 's', 'min', 'hour'."""
...
async def apply(
self, mol: Chem.Mol, config: dict, context: PipelineContext
) -> FilterResult:
"""
Apply this filter to a single molecule.
Args:
mol: RDKit Mol object (with hydrogens if needed)
config: Plugin-specific configuration (validated against config_schema)
context: Shared pipeline context (target, binding site, etc.)
Returns:
FilterResult with passed=True/False, optional score, optional metadata
"""
...
async def apply_batch(
self, mols: list[Chem.Mol], config: dict, context: PipelineContext
) -> list[FilterResult]:
"""
Apply this filter to a batch of molecules.
Default implementation calls apply() for each mol.
Override for batch-optimized implementations (e.g., batch docking).
"""
...
async def setup(self, config: dict, context: PipelineContext) -> None:
"""
Optional one-time setup before processing compounds.
Called once at the start of a pipeline run for this node.
Use for: loading receptor, computing reference fingerprint, etc.
Default: no-op.
"""
...
async def teardown(self) -> None:
"""
Optional cleanup after all compounds are processed.
Default: no-op.
"""
...
Built-in Filters¶
Property Filters (chemlib/plugins/builtin/property_filters.py)¶
| Filter Name | Plugin ID | Description | Key Config | Time |
|---|---|---|---|---|
| Lipinski Rule of Five | lipinski_filter |
MW <= 500, LogP <= 5, HBD <= 5, HBA <= 10 | allow_violations (int, 0-2) |
ms |
| Veber Rules | veber_filter |
TPSA <= 140, RotBonds <= 10 | — | ms |
| Ghose Filter | ghose_filter |
MW 160-480, LogP -0.4-5.6, atoms 20-70, MR 40-130 | — | ms |
| Egan Filter | egan_filter |
TPSA <= 131.6, LogP <= 5.88 | — | ms |
| Muegge Filter | muegge_filter |
MW 200-600, LogP -2-5, TPSA <= 150, rings <= 7, HBD <= 5, HBA <= 10, RotBonds <= 15 | — | ms |
| PAINS Filter | pains_filter |
No PAINS substructures | — | ms |
| Brenk Filter | brenk_filter |
No Brenk structural alerts | — | ms |
| QED Threshold | qed_threshold |
QED score above threshold | threshold (float, 0-1, default 0.5) |
ms |
| SA Score Threshold | sa_threshold |
SA score below threshold (lower = easier) | threshold (float, 1-10, default 5.0) |
ms |
| MW Range | mw_range |
Molecular weight within range | min_mw (float), max_mw (float) |
ms |
| LogP Range | logp_range |
LogP within range | min_logp (float), max_logp (float) |
ms |
| TPSA Range | tpsa_range |
TPSA within range | min_tpsa (float), max_tpsa (float) |
ms |
| HBD Max | hbd_max |
Hydrogen bond donors below max | max_hbd (int) |
ms |
| HBA Max | hba_max |
Hydrogen bond acceptors below max | max_hba (int) |
ms |
| Rotatable Bonds Max | rotbonds_max |
Rotatable bonds below max | max_rotbonds (int) |
ms |
Example Implementation:
class LipinskiFilter:
"""Lipinski Rule of Five filter."""
@property
def name(self) -> str:
return "lipinski_filter"
@property
def display_name(self) -> str:
return "Lipinski Rule of Five"
@property
def category(self) -> str:
return "property"
@property
def description(self) -> str:
return (
"Filters compounds based on Lipinski's Rule of Five: "
"MW <= 500, LogP <= 5, HBD <= 5, HBA <= 10. "
"Configurable number of allowed violations."
)
@property
def config_schema(self) -> dict:
return {
"type": "object",
"properties": {
"allow_violations": {
"type": "integer",
"minimum": 0,
"maximum": 2,
"default": 0,
"title": "Allowed Violations",
"description": "Number of Lipinski rule violations to allow (0-2)"
}
}
}
@property
def estimated_time_per_compound(self) -> str:
return "ms"
async def apply(
self, mol: Chem.Mol, config: dict, context: PipelineContext
) -> FilterResult:
from chemlib.chemistry.filters import check_lipinski
allow = config.get("allow_violations", 0)
result = check_lipinski(mol)
num_violations = len(result["violations"])
passed = num_violations <= allow
return FilterResult(
passed=passed,
score=1.0 - (num_violations / 4.0), # Normalize: 1.0 = perfect, 0.0 = all violated
metadata={
"violations": result["violations"],
"num_violations": num_violations,
"properties": result["properties"],
}
)
async def apply_batch(
self, mols: list[Chem.Mol], config: dict, context: PipelineContext
) -> list[FilterResult]:
return [await self.apply(mol, config, context) for mol in mols]
async def setup(self, config: dict, context: PipelineContext) -> None:
pass
async def teardown(self) -> None:
pass
Similarity Filters (chemlib/plugins/builtin/similarity_filters.py)¶
| Filter Name | Plugin ID | Description | Key Config | Time |
|---|---|---|---|---|
| Morgan FP Tanimoto | tanimoto_similarity |
Tanimoto similarity vs reference compound | reference_smiles (str), threshold (float, 0-1), radius (int, default 2), nbits (int, default 2048) |
ms |
| Substructure Match | substructure_match |
Contains specified SMARTS substructure | smarts (str) |
ms |
| MACCS FP Similarity | maccs_similarity |
MACCS keys Tanimoto vs reference | reference_smiles (str), threshold (float) |
ms |
Tanimoto Filter Implementation Notes:
- In setup(): compute reference fingerprint once, store in context.reference_fp
- In apply(): compute query FP, compute Tanimoto vs stored reference
- Score = Tanimoto coefficient (0-1)
ADME Filters (chemlib/plugins/builtin/adme_filters.py)¶
| Filter Name | Plugin ID | Description | Key Config | Time |
|---|---|---|---|---|
| ESOL Solubility | esol_solubility |
Delaney ESOL aqueous solubility prediction | min_logS (float, default -6.0) |
ms |
| Simple BBB Rule | bbb_rule |
Blood-brain barrier penetration rule (TPSA < 90, MW < 400) | — | ms |
| Simple hERG Rule | herg_rule |
hERG liability rule (LogP > 3.5 AND MW > 400 flags risk) | — | ms |
| Rule of Three | rule_of_three |
Fragment-likeness: MW <= 300, LogP <= 3, HBD <= 3, HBA <= 3, RotBonds <= 3 | — | ms |
Docking Filters (chemlib/plugins/builtin/docking_filter.py)¶
| Filter Name | Plugin ID | Description | Key Config | Time |
|---|---|---|---|---|
| AutoDock Vina | vina_docking |
Dock compound against binding site | binding_site_id (int), exhaustiveness (int, 8-128, default 32), num_poses (int, 1-50, default 10), score_threshold (float, default -6.0 kcal/mol) |
s-min |
| Vina Score Threshold | vina_score_threshold |
Filter by pre-computed docking score | threshold (float, kcal/mol) |
ms |
Vina Docking Filter Implementation Notes:
- In setup(): Prepare receptor PDBQT once, store in context.receptor_pdbqt. Load binding site parameters.
- In apply(): Prepare ligand PDBQT (meeko), run Vina, parse results.
- apply_batch(): Can parallelize docking across compounds.
- Score = Vina binding energy (kcal/mol, more negative = better)
- Metadata includes: poses (PDBQT), interactions (via PLIP on best pose)
- passed = score <= score_threshold
External/API Filters (chemlib/plugins/builtin/external_filters.py)¶
| Filter Name | Plugin ID | Description | Key Config | Time |
|---|---|---|---|---|
| ADMETlab 2.0 | admetlab_api |
Call ADMETlab 2.0 REST API for ADME/Tox | endpoint (str), properties (list[str]) |
s |
| PLIP Interaction Analysis | plip_interaction |
Analyze protein-ligand interactions | min_hbonds (int, default 1), min_hydrophobic (int, default 0) |
s |
Database Schema¶
chemlib/models/pipeline.py¶
from __future__ import annotations
from datetime import datetime
from typing import Optional
from sqlalchemy import String, Text, Integer, Float, Boolean, DateTime, ForeignKey, JSON
from sqlalchemy.orm import Mapped, mapped_column, relationship
from sqlalchemy.sql import func
from chemlib.models.base import Base
class Pipeline(Base):
__tablename__ = "pipelines"
id: Mapped[int] = mapped_column(primary_key=True)
name: Mapped[str] = mapped_column(String(300), nullable=False)
description: Mapped[Optional[str]] = mapped_column(Text, nullable=True)
definition: Mapped[dict] = mapped_column(
JSON, nullable=False # Full PipelineDefinition serialized
)
target_id: Mapped[Optional[int]] = mapped_column(
ForeignKey("protein_targets.id", ondelete="SET NULL"), nullable=True
)
created_at: Mapped[datetime] = mapped_column(
DateTime(timezone=True), server_default=func.now()
)
updated_at: Mapped[datetime] = mapped_column(
DateTime(timezone=True), server_default=func.now(), onupdate=func.now()
)
# Relationships
runs: Mapped[list[PipelineRun]] = relationship(
back_populates="pipeline", cascade="all, delete-orphan"
)
def __repr__(self) -> str:
return f"<Pipeline(id={self.id}, name='{self.name}')>"
class PipelineRun(Base):
__tablename__ = "pipeline_runs"
id: Mapped[int] = mapped_column(primary_key=True)
pipeline_id: Mapped[int] = mapped_column(
ForeignKey("pipelines.id", ondelete="CASCADE"), nullable=False, index=True
)
status: Mapped[str] = mapped_column(
String(20), nullable=False, default="pending"
# "pending", "running", "completed", "failed", "cancelled"
)
total_compounds: Mapped[int] = mapped_column(Integer, nullable=False, default=0)
processed_compounds: Mapped[int] = mapped_column(Integer, nullable=False, default=0)
current_node: Mapped[Optional[str]] = mapped_column(String(100), nullable=True)
started_at: Mapped[Optional[datetime]] = mapped_column(DateTime(timezone=True), nullable=True)
completed_at: Mapped[Optional[datetime]] = mapped_column(DateTime(timezone=True), nullable=True)
summary: Mapped[Optional[dict]] = mapped_column(
JSON, nullable=True
# {"node_1": {"passed": 800, "failed": 200, "total": 1000}, ...}
)
error_message: Mapped[Optional[str]] = mapped_column(Text, nullable=True)
created_at: Mapped[datetime] = mapped_column(
DateTime(timezone=True), server_default=func.now()
)
# Relationships
pipeline: Mapped[Pipeline] = relationship(back_populates="runs")
results: Mapped[list[PipelineRunResult]] = relationship(
back_populates="run", cascade="all, delete-orphan"
)
def __repr__(self) -> str:
return f"<PipelineRun(id={self.id}, status='{self.status}')>"
class PipelineRunResult(Base):
__tablename__ = "pipeline_run_results"
id: Mapped[int] = mapped_column(primary_key=True)
run_id: Mapped[int] = mapped_column(
ForeignKey("pipeline_runs.id", ondelete="CASCADE"), nullable=False, index=True
)
compound_id: Mapped[Optional[int]] = mapped_column(
ForeignKey("compounds.id", ondelete="SET NULL"), nullable=True, index=True
)
assembled_molecule_id: Mapped[Optional[int]] = mapped_column(
ForeignKey("assembled_molecules.id", ondelete="SET NULL"), nullable=True, index=True
)
node_id: Mapped[str] = mapped_column(String(100), nullable=False, index=True)
passed: Mapped[bool] = mapped_column(Boolean, nullable=False)
score: Mapped[Optional[float]] = mapped_column(Float, nullable=True)
metadata: Mapped[Optional[dict]] = mapped_column(JSON, nullable=True)
created_at: Mapped[datetime] = mapped_column(
DateTime(timezone=True), server_default=func.now()
)
# Relationships
run: Mapped[PipelineRun] = relationship(back_populates="results")
# Composite index for efficient queries
__table_args__ = (
# Index for "get all results for a specific run + node"
# Already covered by individual indexes, but composite may help
)
def __repr__(self) -> str:
return f"<PipelineRunResult(run={self.run_id}, node='{self.node_id}', passed={self.passed})>"
class FilterPluginRegistry(Base):
__tablename__ = "filter_plugin_registry"
id: Mapped[int] = mapped_column(primary_key=True)
name: Mapped[str] = mapped_column(String(100), unique=True, nullable=False)
display_name: Mapped[str] = mapped_column(String(200), nullable=False)
category: Mapped[str] = mapped_column(
String(30), nullable=False, index=True
# "property", "similarity", "docking", "adme", "external"
)
description: Mapped[Optional[str]] = mapped_column(Text, nullable=True)
plugin_class: Mapped[str] = mapped_column(
String(300), nullable=False
# Dotted path: "chemlib.plugins.builtin.property_filters.LipinskiFilter"
)
config_schema: Mapped[Optional[dict]] = mapped_column(JSON, nullable=True)
estimated_time: Mapped[Optional[str]] = mapped_column(String(10), nullable=True) # "ms", "s", "min"
is_builtin: Mapped[bool] = mapped_column(Boolean, default=True)
is_active: Mapped[bool] = mapped_column(Boolean, default=True)
version: Mapped[str] = mapped_column(String(20), nullable=False, default="1.0.0")
created_at: Mapped[datetime] = mapped_column(
DateTime(timezone=True), server_default=func.now()
)
def __repr__(self) -> str:
return f"<FilterPluginRegistry(name='{self.name}', category='{self.category}')>"
Pipeline Executor¶
chemlib/services/pipeline_executor.py¶
"""
Pipeline executor — processes compounds through a DAG of filter nodes.
The executor:
1. Validates the pipeline DAG (no cycles, all plugins exist)
2. Performs topological sort to determine execution order
3. Processes compounds in batches through each node
4. Tracks progress and stores results in the database
5. Supports early termination (if a compound fails, skip downstream nodes)
6. Runs as a background task for long pipelines
"""
from __future__ import annotations
import asyncio
import importlib
from collections import defaultdict
from typing import Any
from sqlalchemy.ext.asyncio import AsyncSession
from rdkit import Chem
from chemlib.plugins.protocols import FilterPlugin, FilterResult, PipelineContext
from chemlib.schemas.pipeline import PipelineDefinition, PipelineNode, PipelineEdge
class PipelineExecutor:
"""Executes a screening pipeline against a set of compounds."""
def __init__(self, db: AsyncSession, pipeline_run_id: int):
self.db = db
self.run_id = pipeline_run_id
self._plugins: dict[str, FilterPlugin] = {} # node_id -> plugin instance
self._node_map: dict[str, PipelineNode] = {}
self._adjacency: dict[str, list[tuple[str, str]]] = {} # node_id -> [(target_id, condition)]
self._reverse_adj: dict[str, list[tuple[str, str]]] = {} # node_id -> [(source_id, condition)]
async def execute(
self,
definition: PipelineDefinition,
compounds: list[dict], # [{"id": 1, "smiles": "CCO", "source": "compound"}, ...]
batch_size: int = 100
) -> None:
"""
Main execution method.
Steps:
1. Build DAG data structures (adjacency lists)
2. Topological sort for execution order
3. Instantiate plugin objects for each node
4. Create PipelineContext with shared state
5. Call setup() on all plugins
6. For each node in topological order:
a. Determine which compounds reach this node
(based on incoming edges and their conditions)
b. Process compounds in batches through the plugin
c. Store FilterResult per compound per node
d. Update PipelineRun progress
7. Call teardown() on all plugins
8. Update PipelineRun to "completed"
"""
async def _build_dag(self, definition: PipelineDefinition) -> list[str]:
"""
Build adjacency lists and return topological order.
Uses Kahn's algorithm:
1. Compute in-degree for each node
2. Initialize queue with nodes having in-degree 0
3. Process queue: for each node, reduce in-degree of successors
4. If all nodes processed, return order; else raise CycleError
"""
self._node_map = {node.id: node for node in definition.nodes}
# Build adjacency
self._adjacency = defaultdict(list)
self._reverse_adj = defaultdict(list)
in_degree = defaultdict(int)
for node in definition.nodes:
in_degree.setdefault(node.id, 0)
for edge in definition.edges:
self._adjacency[edge.source_node_id].append(
(edge.target_node_id, edge.condition)
)
self._reverse_adj[edge.target_node_id].append(
(edge.source_node_id, edge.condition)
)
in_degree[edge.target_node_id] += 1
# Kahn's algorithm
queue = [nid for nid, deg in in_degree.items() if deg == 0]
order = []
while queue:
nid = queue.pop(0)
order.append(nid)
for target_id, _ in self._adjacency.get(nid, []):
in_degree[target_id] -= 1
if in_degree[target_id] == 0:
queue.append(target_id)
if len(order) != len(definition.nodes):
raise PipelineError("Pipeline contains a cycle")
return order
async def _instantiate_plugin(self, node: PipelineNode) -> FilterPlugin:
"""
Load and instantiate a plugin from the registry.
1. Look up plugin_id in FilterPluginRegistry table
2. Import the plugin_class (dotted path)
3. Instantiate and return
"""
# Example: "chemlib.plugins.builtin.property_filters.LipinskiFilter"
module_path, class_name = node.plugin_id.rsplit(".", 1)
module = importlib.import_module(module_path)
plugin_class = getattr(module, class_name)
return plugin_class()
async def _get_compounds_for_node(
self,
node_id: str,
compound_results: dict[int, dict[str, FilterResult]]
) -> list[dict]:
"""
Determine which compounds should flow into this node.
For each incoming edge (source_node_id, condition):
- If condition == "pass": include compounds that passed source node
- If condition == "fail": include compounds that failed source node
- If condition == "all": include all compounds that reached source node
For source nodes (no incoming edges): include all initial compounds.
Take the UNION of compounds from all incoming edges.
"""
async def _process_batch(
self,
plugin: FilterPlugin,
mols: list[tuple[dict, Chem.Mol]], # (compound_info, mol) pairs
config: dict,
context: PipelineContext
) -> list[tuple[dict, FilterResult]]:
"""
Process a batch of molecules through a plugin.
Returns list of (compound_info, result) pairs.
"""
mol_objects = [mol for _, mol in mols]
results = await plugin.apply_batch(mol_objects, config, context)
return [(info, result) for (info, _), result in zip(mols, results)]
async def _store_results(
self,
node_id: str,
results: list[tuple[dict, FilterResult]]
) -> None:
"""
Store FilterResults in PipelineRunResult table.
Batch insert for efficiency.
"""
async def _update_progress(
self,
node_id: str,
passed: int,
failed: int,
total: int
) -> None:
"""Update PipelineRun with current progress and summary."""
async def cancel(self) -> None:
"""Cancel a running pipeline. Sets status to 'cancelled'."""
Execution Flow Diagram¶
execute() called
│
├── _build_dag() → topological order: [source, lipinski, pains, qed, similarity, docking, sink]
│
├── Instantiate plugins for each non-source/sink node
│
├── Create PipelineContext (load target, binding site, etc.)
│
├── Call setup() on all plugins
│
├── For node "lipinski" (first filter):
│ ├── _get_compounds_for_node("lipinski") → all 1000 compounds
│ ├── Convert SMILES to Mol objects
│ ├── Batch 1 (100 cpds): _process_batch() → [FilterResult, ...]
│ ├── Batch 2 (100 cpds): _process_batch() → [FilterResult, ...]
│ ├── ... (10 batches)
│ ├── _store_results("lipinski", results)
│ └── _update_progress("lipinski", passed=800, failed=200, total=1000)
│
├── For node "pains":
│ ├── _get_compounds_for_node("pains") → 800 compounds (lipinski passed)
│ ├── Process in batches...
│ └── _update_progress("pains", passed=750, failed=50, total=800)
│
├── ... (continue through all nodes)
│
├── Call teardown() on all plugins
│
└── Update PipelineRun status → "completed"
Service Layer¶
chemlib/services/pipeline_service.py¶
class PipelineService:
"""Business logic for pipeline definition management."""
async def create_pipeline(
self, db: AsyncSession, data: PipelineCreate
) -> PipelineResponse:
"""
Create a new pipeline definition.
Steps:
1. Validate the DAG (no cycles, all plugin_ids exist in registry)
2. Validate each node's config against its plugin's config_schema
3. Persist Pipeline record with definition as JSON
"""
async def update_pipeline(
self, db: AsyncSession, pipeline_id: int, data: PipelineCreate
) -> PipelineResponse:
"""Update an existing pipeline definition."""
async def get(self, db: AsyncSession, pipeline_id: int) -> PipelineResponse | None:
"""Get pipeline by ID."""
async def list_pipelines(
self, db: AsyncSession, limit: int = 50, offset: int = 0
) -> PipelineListResponse:
"""List all pipelines with pagination."""
async def delete(self, db: AsyncSession, pipeline_id: int) -> bool:
"""Delete a pipeline and all associated runs."""
async def start_run(
self, db: AsyncSession, pipeline_id: int, data: PipelineRunCreate
) -> PipelineRunResponse:
"""
Start a pipeline run.
Steps:
1. Load pipeline definition
2. Resolve compound list based on data (specific IDs or all)
3. Create PipelineRun record with status="pending"
4. Spawn background task: _execute_pipeline_background()
5. Return run response immediately (async execution)
"""
async def _execute_pipeline_background(
self, run_id: int, pipeline_id: int, compound_list: list[dict]
) -> None:
"""
Background task that creates a PipelineExecutor and runs the pipeline.
Catches exceptions, updates PipelineRun status to "failed" on error.
"""
async def get_run(self, db: AsyncSession, run_id: int) -> PipelineRunResponse | None:
"""Get pipeline run status."""
async def get_run_results(
self, db: AsyncSession, run_id: int, filters: PipelineRunResultFilter
) -> list[PipelineRunResultResponse]:
"""Get results for a pipeline run with filtering and pagination."""
async def cancel_run(self, db: AsyncSession, run_id: int) -> PipelineRunResponse:
"""Cancel a running pipeline."""
async def get_funnel_summary(
self, db: AsyncSession, run_id: int
) -> list[dict]:
"""
Get funnel visualization data for a pipeline run.
Returns: [
{"node_id": "lipinski", "label": "Lipinski RO5", "total": 1000, "passed": 800, "failed": 200},
{"node_id": "pains", "label": "PAINS Filter", "total": 800, "passed": 750, "failed": 50},
...
]
Ordered by execution order.
"""
API Endpoints¶
Pipelines — chemlib/api/pipelines.py¶
| Method | Endpoint | Description | Request Body | Response |
|---|---|---|---|---|
GET |
/api/pipelines/ |
List all pipelines | Query: limit, offset | PipelineListResponse |
POST |
/api/pipelines/ |
Create a pipeline | PipelineCreate | PipelineResponse |
GET |
/api/pipelines/{id} |
Get pipeline details | — | PipelineResponse |
PUT |
/api/pipelines/{id} |
Update pipeline | PipelineCreate | PipelineResponse |
DELETE |
/api/pipelines/{id} |
Delete pipeline + runs | — | 204 |
POST |
/api/pipelines/{id}/run |
Start a pipeline run | PipelineRunCreate | PipelineRunResponse |
GET |
/api/pipelines/runs/{run_id} |
Get run status | — | PipelineRunResponse |
GET |
/api/pipelines/runs/{run_id}/results |
Get run results | Query: PipelineRunResultFilter | list[PipelineRunResultResponse] |
GET |
/api/pipelines/runs/{run_id}/funnel |
Get funnel summary | — | list[dict] |
POST |
/api/pipelines/runs/{run_id}/cancel |
Cancel a run | — | PipelineRunResponse |
Plugins — chemlib/api/plugins.py¶
| Method | Endpoint | Description | Response |
|---|---|---|---|
GET |
/api/plugins/ |
List all registered plugins | list[PluginRegistryResponse] |
GET |
/api/plugins/{name} |
Get plugin details + config schema | PluginRegistryResponse |
GET |
/api/plugins/categories |
List available categories | list[str] |
GET |
/api/plugins/category/{category} |
List plugins by category | list[PluginRegistryResponse] |
class PluginRegistryResponse(BaseModel):
id: int
name: str
display_name: str
category: str
description: str | None
config_schema: dict | None
estimated_time: str | None
is_builtin: bool
is_active: bool
version: str
model_config = ConfigDict(from_attributes=True)
UI: Pipeline Builder¶
Pipeline Builder Page (pipeline_builder.html)¶
The pipeline builder is a visual DAG editor where users drag-and-drop filter nodes onto a canvas, connect them with edges, and configure each node.
┌─────────────────────────────────────────────────────────────────────────┐
│ Pipeline Builder: [Pipeline Name Input] [Save] [Run] │
├─────────────────────────────────────────────────────────────────────────┤
│ │
│ ┌──────────────┐ ┌───────────────────────────────────────────────┐ │
│ │ Filter Nodes │ │ │ │
│ │ (Drag & Drop)│ │ CANVAS (DAG Editor) │ │
│ │ │ │ │ │
│ │ ▸ Property │ │ ┌────────────┐ │ │
│ │ □ Lipinski │ │ │ Library │ │ │
│ │ □ PAINS │ │ │ (Source) │ │ │
│ │ □ QED │ │ └─────┬──────┘ │ │
│ │ □ MW Range │ │ │ all │ │
│ │ □ LogP │ │ ▼ │ │
│ │ │ │ ┌────────────┐ │ │
│ │ ▸ Similarity │ │ │ Lipinski │ ← click to configure │ │
│ │ □ Tanimoto │ │ │ RO5 │ [allow_violations: 1] │ │
│ │ □ Substruct│ │ └─────┬──────┘ │ │
│ │ □ MACCS │ │ │ pass │ │
│ │ │ │ ▼ │ │
│ │ ▸ ADME │ │ ┌────────────┐ │ │
│ │ □ ESOL │ │ │ PAINS │ │ │
│ │ □ BBB │ │ │ Filter │ │ │
│ │ │ │ └─────┬──────┘ │ │
│ │ ▸ Docking │ │ │ pass │ │
│ │ □ Vina │ │ ▼ │ │
│ │ │ │ ┌────────────┐ │ │
│ │ ▸ External │ │ │ Vina Dock │ │ │
│ │ □ ADMETlab │ │ │ (site:1) │ │ │
│ │ □ PLIP │ │ └─────┬──────┘ │ │
│ │ │ │ │ pass │ │
│ └──────────────┘ │ ▼ │ │
│ │ ┌────────────┐ │ │
│ ┌──────────────┐ │ │ Results │ │ │
│ │ Node Config │ │ │ (Sink) │ │ │
│ │ │ │ └────────────┘ │ │
│ │ Lipinski RO5 │ │ │ │
│ │ ──────────── │ └───────────────────────────────────────────────┘ │
│ │ Allow │ │
│ │ violations: 1 │ │
│ │ │ │
│ │ [Apply] │ │
│ └──────────────┘ │
└─────────────────────────────────────────────────────────────────────────┘
Implementation: chemlib/static/js/pipeline_editor.js
The DAG editor can be implemented using a lightweight canvas library or SVG. Key interactions:
- Drag node from sidebar onto canvas → creates a PipelineNode
- Connect nodes by dragging from output port to input port → creates a PipelineEdge
- Click node → opens config panel on the side (auto-generated from config_schema)
- Delete node → removes node and connected edges
- Save → serializes canvas to PipelineDefinition JSON, POSTs to /api/pipelines/
- Run → POSTs to /api/pipelines/{id}/run
Pipeline Results Page (pipeline_results.html)¶
┌─────────────────────────────────────────────────────────────────────────┐
│ Pipeline Run: "EGFR Screening v2" Status: ✓ Completed │
│ Started: 2026-03-27 10:00 Completed: 2026-03-27 10:45 │
│ Total compounds: 5,000 │
├─────────────────────────────────────────────────────────────────────────┤
│ │
│ FUNNEL VISUALIZATION │
│ │
│ ┌─────────────────────────────────────────────────────────────┐ │
│ │ Library Input ████████████████████ 5,000 │ │
│ │ Lipinski RO5 ██████████████████ 4,200 │ │
│ │ PAINS Filter █████████████████ 3,900 │ │
│ │ QED > 0.4 ████████████ 2,100 │ │
│ │ Tanimoto > 0.3 █████████ 1,500 │ │
│ │ Vina Docking (< -6.0) ████ 320 │ │
│ │ PLIP (>= 2 H-bonds) ██ 120 │ │
│ └─────────────────────────────────────────────────────────────┘ │
│ │
│ RESULTS TABLE (120 compounds survived) [Export CSV] [Export SDF]│
│ │
│ Filter by stage: [All ▾] Sort: [Docking Score ▾] [Ascending ▾] │
│ │
│ ┌──────┬───────────────────┬─────────┬──────────┬───────────┬───────┐ │
│ │ # │ Compound │ Dock │ QED │ H-bonds │ View │ │
│ ├──────┼───────────────────┼─────────┼──────────┼───────────┼───────┤ │
│ │ 1 │ CPD-00234 │ -9.2 │ 0.78 │ 4 │ [3D] │ │
│ │ 2 │ ASM-00056 │ -8.8 │ 0.71 │ 3 │ [3D] │ │
│ │ 3 │ CPD-01102 │ -8.5 │ 0.65 │ 3 │ [3D] │ │
│ │ ... │ │
│ └──────┴───────────────────┴─────────┴──────────┴───────────┴───────┘ │
│ ◀ 1 2 3 ... ▶ │
│ │
│ [Click "3D" to see docked pose in protein viewer] │
└─────────────────────────────────────────────────────────────────────────┘
Background Execution¶
Long-running pipelines (especially those with docking) are executed as background tasks using FastAPI's BackgroundTasks or an async task queue.
Approach 1: FastAPI BackgroundTasks (Simple)¶
@router.post("/{pipeline_id}/run")
async def start_pipeline_run(
pipeline_id: int,
data: PipelineRunCreate,
background_tasks: BackgroundTasks,
db: AsyncSession = Depends(get_db),
service: PipelineService = Depends(),
):
run = await service.create_run(db, pipeline_id, data)
background_tasks.add_task(service._execute_pipeline_background, run.id, pipeline_id)
return run
Approach 2: Async Task Queue (Future — for scaling)¶
For production with many concurrent users, replace BackgroundTasks with a proper task queue (e.g., Celery with Redis, or arq). The API remains the same; only the dispatching mechanism changes.
Progress Polling¶
The UI polls /api/pipelines/runs/{run_id} every few seconds to check progress:
async function pollRunStatus(runId) {
const response = await fetch(`/api/pipelines/runs/${runId}`);
const data = await response.json();
updateProgressBar(data.processed_compounds, data.total_compounds);
updateCurrentStep(data.current_node);
if (data.status === "running") {
setTimeout(() => pollRunStatus(runId), 3000); // Poll every 3 seconds
} else if (data.status === "completed") {
loadResults(runId);
} else if (data.status === "failed") {
showError(data.error_message);
}
}
Seeding Built-in Plugins¶
scripts/seed_plugins.py¶
"""Seed the FilterPluginRegistry with all built-in plugins."""
BUILTIN_PLUGINS = [
{
"name": "lipinski_filter",
"display_name": "Lipinski Rule of Five",
"category": "property",
"description": "Filters based on Lipinski's RO5: MW<=500, LogP<=5, HBD<=5, HBA<=10",
"plugin_class": "chemlib.plugins.builtin.property_filters.LipinskiFilter",
"config_schema": {
"type": "object",
"properties": {
"allow_violations": {
"type": "integer", "minimum": 0, "maximum": 2,
"default": 0, "title": "Allowed Violations"
}
}
},
"estimated_time": "ms",
"is_builtin": True,
"version": "1.0.0",
},
{
"name": "veber_filter",
"display_name": "Veber Rules",
"category": "property",
"description": "Veber oral bioavailability rules: TPSA<=140, RotBonds<=10",
"plugin_class": "chemlib.plugins.builtin.property_filters.VeberFilter",
"config_schema": {"type": "object", "properties": {}},
"estimated_time": "ms",
"is_builtin": True,
"version": "1.0.0",
},
# ... (all other built-in plugins defined similarly)
{
"name": "pains_filter",
"display_name": "PAINS Filter",
"category": "property",
"description": "Pan-Assay Interference Compounds (PAINS) structural alert filter",
"plugin_class": "chemlib.plugins.builtin.property_filters.PAINSFilter",
"config_schema": {"type": "object", "properties": {}},
"estimated_time": "ms",
"is_builtin": True,
"version": "1.0.0",
},
{
"name": "qed_threshold",
"display_name": "QED Score Threshold",
"category": "property",
"description": "Filter by Quantitative Estimate of Drug-likeness (QED) score",
"plugin_class": "chemlib.plugins.builtin.property_filters.QEDThresholdFilter",
"config_schema": {
"type": "object",
"properties": {
"threshold": {
"type": "number", "minimum": 0, "maximum": 1,
"default": 0.5, "title": "Minimum QED Score"
}
}
},
"estimated_time": "ms",
"is_builtin": True,
"version": "1.0.0",
},
{
"name": "tanimoto_similarity",
"display_name": "Morgan FP Tanimoto Similarity",
"category": "similarity",
"description": "Filter by Tanimoto similarity (Morgan/ECFP fingerprint) to a reference compound",
"plugin_class": "chemlib.plugins.builtin.similarity_filters.TanimotoSimilarityFilter",
"config_schema": {
"type": "object",
"properties": {
"reference_smiles": {
"type": "string", "title": "Reference SMILES",
"description": "SMILES of the reference compound"
},
"threshold": {
"type": "number", "minimum": 0, "maximum": 1,
"default": 0.5, "title": "Minimum Tanimoto Similarity"
},
"radius": {
"type": "integer", "minimum": 1, "maximum": 4,
"default": 2, "title": "Morgan FP Radius"
},
"nbits": {
"type": "integer", "enum": [1024, 2048, 4096],
"default": 2048, "title": "Fingerprint Bits"
}
},
"required": ["reference_smiles"]
},
"estimated_time": "ms",
"is_builtin": True,
"version": "1.0.0",
},
{
"name": "vina_docking",
"display_name": "AutoDock Vina Docking",
"category": "docking",
"description": "Dock compound against a protein binding site using AutoDock Vina",
"plugin_class": "chemlib.plugins.builtin.docking_filter.VinaDockingFilter",
"config_schema": {
"type": "object",
"properties": {
"binding_site_id": {
"type": "integer", "title": "Binding Site",
"description": "ID of the binding site to dock against"
},
"exhaustiveness": {
"type": "integer", "minimum": 8, "maximum": 128,
"default": 32, "title": "Exhaustiveness"
},
"num_poses": {
"type": "integer", "minimum": 1, "maximum": 50,
"default": 10, "title": "Number of Poses"
},
"score_threshold": {
"type": "number", "maximum": 0,
"default": -6.0, "title": "Score Threshold (kcal/mol)",
"description": "Compounds with score <= threshold pass"
}
},
"required": ["binding_site_id"]
},
"estimated_time": "min",
"is_builtin": True,
"version": "1.0.0",
},
]
async def seed_plugins(db: AsyncSession):
"""Insert all built-in plugins into the registry."""
for plugin_data in BUILTIN_PLUGINS:
existing = await db.execute(
select(FilterPluginRegistry).where(
FilterPluginRegistry.name == plugin_data["name"]
)
)
if existing.scalar_one_or_none() is None:
db.add(FilterPluginRegistry(**plugin_data))
await db.commit()
Alembic Migration¶
# alembic/versions/xxxx_add_pipeline_tables.py
def upgrade():
op.create_table(
"filter_plugin_registry",
sa.Column("id", sa.Integer, primary_key=True),
sa.Column("name", sa.String(100), unique=True, nullable=False),
sa.Column("display_name", sa.String(200), nullable=False),
sa.Column("category", sa.String(30), nullable=False, index=True),
sa.Column("description", sa.Text, nullable=True),
sa.Column("plugin_class", sa.String(300), nullable=False),
sa.Column("config_schema", sa.JSON, nullable=True),
sa.Column("estimated_time", sa.String(10), nullable=True),
sa.Column("is_builtin", sa.Boolean, default=True),
sa.Column("is_active", sa.Boolean, default=True),
sa.Column("version", sa.String(20), nullable=False, default="1.0.0"),
sa.Column("created_at", sa.DateTime(timezone=True), server_default=sa.func.now()),
)
op.create_table(
"pipelines",
sa.Column("id", sa.Integer, primary_key=True),
sa.Column("name", sa.String(300), nullable=False),
sa.Column("description", sa.Text, nullable=True),
sa.Column("definition", sa.JSON, nullable=False),
sa.Column("target_id", sa.Integer, sa.ForeignKey("protein_targets.id", ondelete="SET NULL"), nullable=True),
sa.Column("created_at", sa.DateTime(timezone=True), server_default=sa.func.now()),
sa.Column("updated_at", sa.DateTime(timezone=True), server_default=sa.func.now()),
)
op.create_table(
"pipeline_runs",
sa.Column("id", sa.Integer, primary_key=True),
sa.Column("pipeline_id", sa.Integer, sa.ForeignKey("pipelines.id", ondelete="CASCADE"), nullable=False, index=True),
sa.Column("status", sa.String(20), nullable=False, default="pending"),
sa.Column("total_compounds", sa.Integer, nullable=False, default=0),
sa.Column("processed_compounds", sa.Integer, nullable=False, default=0),
sa.Column("current_node", sa.String(100), nullable=True),
sa.Column("started_at", sa.DateTime(timezone=True), nullable=True),
sa.Column("completed_at", sa.DateTime(timezone=True), nullable=True),
sa.Column("summary", sa.JSON, nullable=True),
sa.Column("error_message", sa.Text, nullable=True),
sa.Column("created_at", sa.DateTime(timezone=True), server_default=sa.func.now()),
)
op.create_table(
"pipeline_run_results",
sa.Column("id", sa.Integer, primary_key=True),
sa.Column("run_id", sa.Integer, sa.ForeignKey("pipeline_runs.id", ondelete="CASCADE"), nullable=False, index=True),
sa.Column("compound_id", sa.Integer, sa.ForeignKey("compounds.id", ondelete="SET NULL"), nullable=True, index=True),
sa.Column("assembled_molecule_id", sa.Integer, sa.ForeignKey("assembled_molecules.id", ondelete="SET NULL"), nullable=True, index=True),
sa.Column("node_id", sa.String(100), nullable=False, index=True),
sa.Column("passed", sa.Boolean, nullable=False),
sa.Column("score", sa.Float, nullable=True),
sa.Column("metadata", sa.JSON, nullable=True),
sa.Column("created_at", sa.DateTime(timezone=True), server_default=sa.func.now()),
)
def downgrade():
op.drop_table("pipeline_run_results")
op.drop_table("pipeline_runs")
op.drop_table("pipelines")
op.drop_table("filter_plugin_registry")