Protein Target Library — Design Document¶
Overview¶
The Protein Target Library module enables users to import, browse, and manage protein targets and their 3D structures. It integrates with external databases (UniProt, RCSB PDB, AlphaFold DB) to fetch sequence and structural data, and provides tools for binding site detection, sequence alignment, and structural alignment.
This module is the foundation for all downstream drug discovery tasks: you need a protein target before you can dock compounds against it.
Click diagram to zoom and pan:
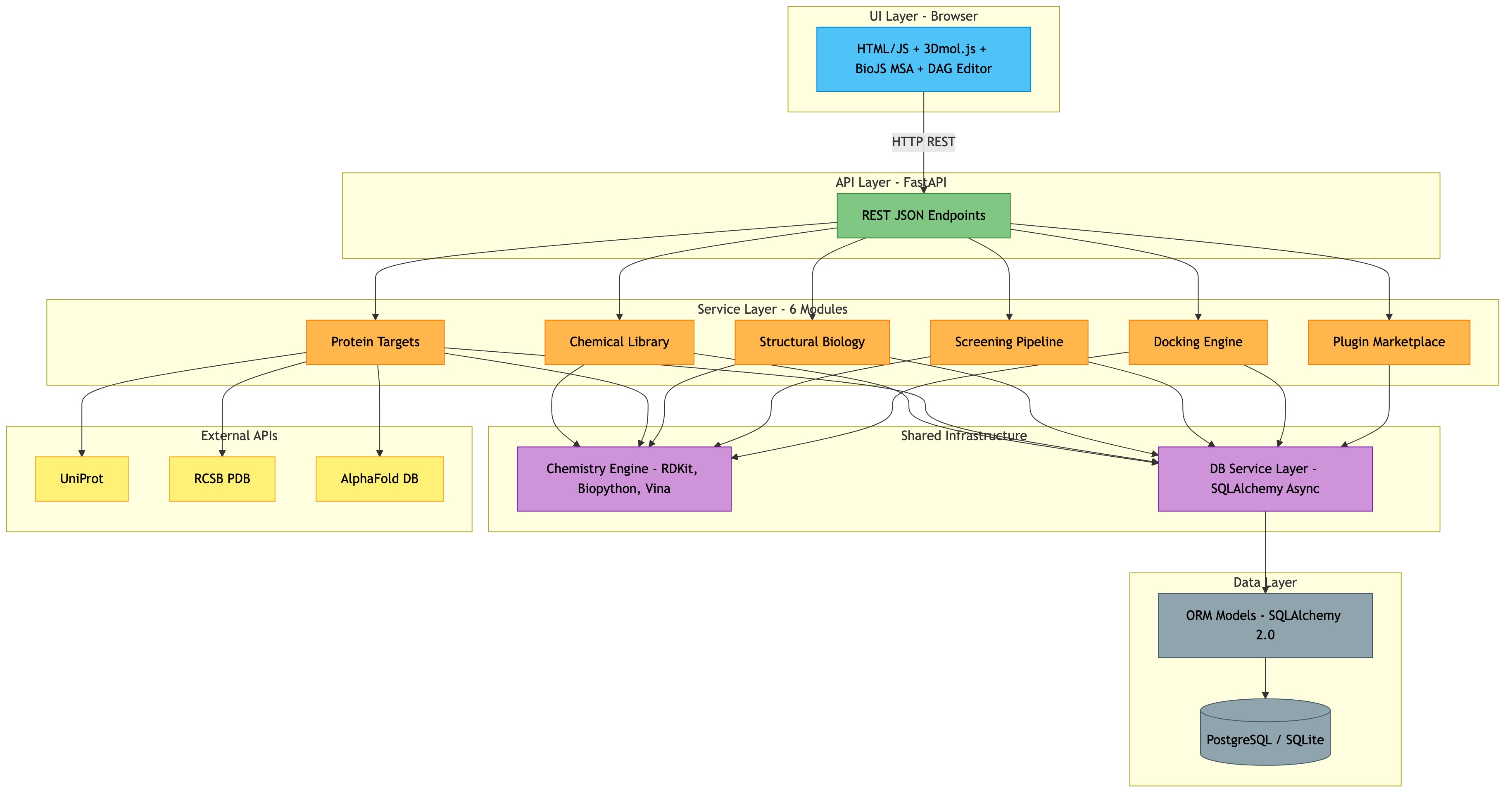
Database Schema¶
Click diagram to zoom and pan:
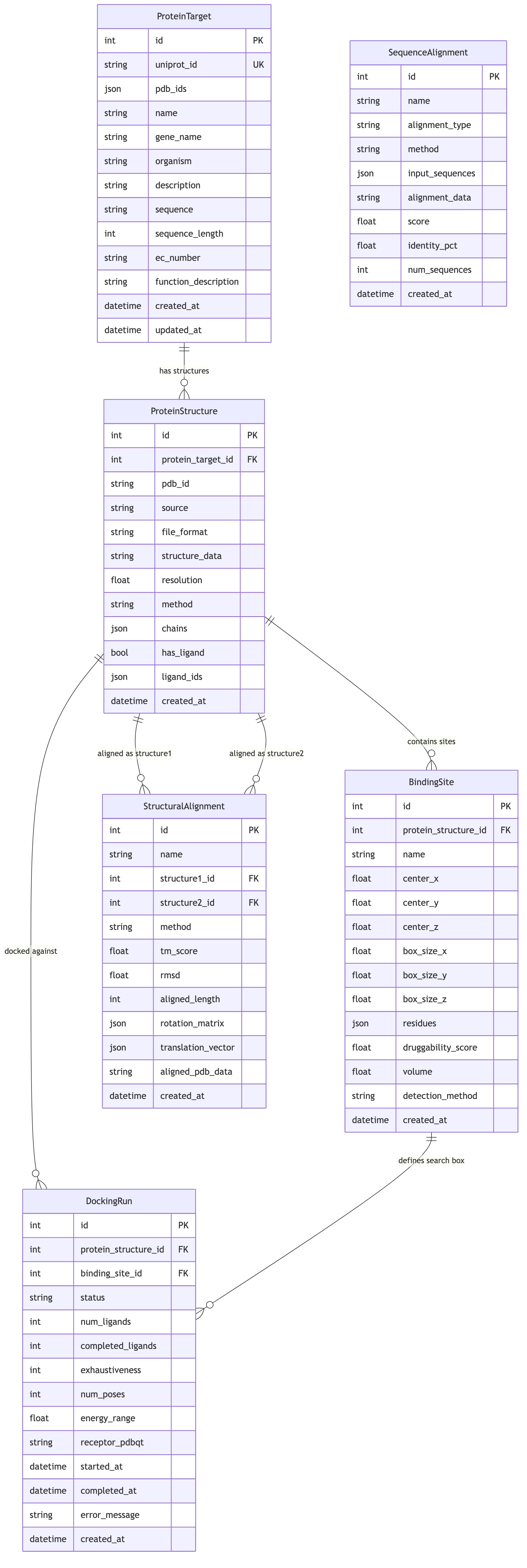
Entity-Relationship Diagram¶
┌────────────────────┐ ┌───────────────────────┐
│ ProteinTarget │ │ ProteinStructure │
├────────────────────┤ ├───────────────────────┤
│ id (PK) │ │ id (PK) │
│ uniprot_id (UQ,IX) │◀──┐ │ protein_target_id (FK)│──▶ ProteinTarget.id
│ pdb_ids (JSON) │ │ │ pdb_id │
│ name │ │ │ source │ (rcsb/alphafold/user_upload)
│ gene_name │ │ │ file_format │ (pdb/mmcif)
│ organism │ │ │ structure_data (Text) │ (full PDB/mmCIF content)
│ description (Text) │ │ │ resolution │
│ sequence (Text) │ │ │ method │ (xray/nmr/cryo_em/predicted)
│ sequence_length │ │ │ chains (JSON) │
│ ec_number │ │ │ has_ligand │
│ function_desc(Text)│ │ │ ligand_ids (JSON) │
│ created_at │ │ │ created_at │
│ updated_at │ │ └───────────────────────┘
└────────────────────┘ │
│ 1:N
▼
┌───────────────────────┐
│ BindingSite │
├───────────────────────┤
│ id (PK) │
│ protein_structure_id │──▶ ProteinStructure.id
│ name │
│ center_x (Float) │
│ center_y (Float) │
│ center_z (Float) │
│ box_size_x (Float) │
│ box_size_y (Float) │
│ box_size_z (Float) │
│ residues (JSON) │ list of "chain:resnum"
│ druggability_score │
│ volume (Float) │
│ detection_method │ (fpocket/manual/ligand_based)
│ created_at │
└───────────────────────┘
┌───────────────────────────┐
│ SequenceAlignment │
├───────────────────────────┤
│ id (PK) │
│ name │
│ alignment_type │ (pairwise/multiple)
│ method │ (biopython/mafft/clustalo)
│ input_sequences (JSON) │ list of {id, name, sequence}
│ alignment_data (Text) │ aligned FASTA format
│ score (Float, nullable) │
│ identity_pct (Float, null)│
│ num_sequences (Int) │
│ created_at │
└───────────────────────────┘
┌───────────────────────────┐
│ StructuralAlignment │
├───────────────────────────┤
│ id (PK) │
│ name │
│ structure1_id (FK) │──▶ ProteinStructure.id
│ structure2_id (FK) │──▶ ProteinStructure.id
│ method │ (tmalign/superimposer)
│ tm_score (Float) │
│ rmsd (Float) │
│ aligned_length (Int) │
│ rotation_matrix (JSON) │ 3x3 matrix as nested list
│ translation_vector (JSON) │ [x, y, z]
│ aligned_pdb_data (Text) │ transformed PDB for overlay
│ created_at │
└───────────────────────────┘
ORM Models¶
chemlib/models/protein.py¶
from __future__ import annotations
from datetime import datetime
from typing import Optional
from sqlalchemy import String, Text, Integer, Float, Boolean, DateTime, ForeignKey, JSON, Index
from sqlalchemy.orm import Mapped, mapped_column, relationship
from sqlalchemy.sql import func
from chemlib.models.base import Base
class ProteinTarget(Base):
__tablename__ = "protein_targets"
id: Mapped[int] = mapped_column(primary_key=True)
uniprot_id: Mapped[Optional[str]] = mapped_column(
String(20), unique=True, index=True, nullable=True
)
pdb_ids: Mapped[Optional[list]] = mapped_column(JSON, nullable=True) # ["1M17", "3W2S"]
name: Mapped[str] = mapped_column(String(500), nullable=False)
gene_name: Mapped[Optional[str]] = mapped_column(String(100), nullable=True)
organism: Mapped[Optional[str]] = mapped_column(String(200), nullable=True)
description: Mapped[Optional[str]] = mapped_column(Text, nullable=True)
sequence: Mapped[Optional[str]] = mapped_column(Text, nullable=True)
sequence_length: Mapped[Optional[int]] = mapped_column(Integer, nullable=True)
ec_number: Mapped[Optional[str]] = mapped_column(String(50), nullable=True)
function_description: Mapped[Optional[str]] = mapped_column(Text, nullable=True)
created_at: Mapped[datetime] = mapped_column(
DateTime(timezone=True), server_default=func.now()
)
updated_at: Mapped[datetime] = mapped_column(
DateTime(timezone=True), server_default=func.now(), onupdate=func.now()
)
# Relationships
structures: Mapped[list[ProteinStructure]] = relationship(
back_populates="protein_target", cascade="all, delete-orphan"
)
def __repr__(self) -> str:
return f"<ProteinTarget(id={self.id}, name='{self.name}', uniprot_id='{self.uniprot_id}')>"
class ProteinStructure(Base):
__tablename__ = "protein_structures"
id: Mapped[int] = mapped_column(primary_key=True)
protein_target_id: Mapped[int] = mapped_column(
ForeignKey("protein_targets.id", ondelete="CASCADE"), nullable=False, index=True
)
pdb_id: Mapped[Optional[str]] = mapped_column(String(10), nullable=True, index=True)
source: Mapped[str] = mapped_column(
String(20), nullable=False # "rcsb", "alphafold", "user_upload"
)
file_format: Mapped[str] = mapped_column(
String(10), nullable=False, default="pdb" # "pdb", "mmcif"
)
structure_data: Mapped[str] = mapped_column(
Text, nullable=False # Full PDB or mmCIF file content
)
resolution: Mapped[Optional[float]] = mapped_column(Float, nullable=True)
method: Mapped[Optional[str]] = mapped_column(
String(20), nullable=True # "xray", "nmr", "cryo_em", "predicted"
)
chains: Mapped[Optional[list]] = mapped_column(JSON, nullable=True) # ["A", "B"]
has_ligand: Mapped[bool] = mapped_column(Boolean, default=False)
ligand_ids: Mapped[Optional[list]] = mapped_column(JSON, nullable=True) # ["ATP", "MG"]
created_at: Mapped[datetime] = mapped_column(
DateTime(timezone=True), server_default=func.now()
)
# Relationships
protein_target: Mapped[ProteinTarget] = relationship(back_populates="structures")
binding_sites: Mapped[list[BindingSite]] = relationship(
back_populates="protein_structure", cascade="all, delete-orphan"
)
def __repr__(self) -> str:
return f"<ProteinStructure(id={self.id}, pdb_id='{self.pdb_id}', source='{self.source}')>"
class BindingSite(Base):
__tablename__ = "binding_sites"
id: Mapped[int] = mapped_column(primary_key=True)
protein_structure_id: Mapped[int] = mapped_column(
ForeignKey("protein_structures.id", ondelete="CASCADE"), nullable=False, index=True
)
name: Mapped[str] = mapped_column(String(200), nullable=False)
center_x: Mapped[float] = mapped_column(Float, nullable=False)
center_y: Mapped[float] = mapped_column(Float, nullable=False)
center_z: Mapped[float] = mapped_column(Float, nullable=False)
box_size_x: Mapped[float] = mapped_column(Float, nullable=False, default=20.0)
box_size_y: Mapped[float] = mapped_column(Float, nullable=False, default=20.0)
box_size_z: Mapped[float] = mapped_column(Float, nullable=False, default=20.0)
residues: Mapped[Optional[list]] = mapped_column(
JSON, nullable=True # ["A:GLU45", "A:ASP52", "A:LYS721"]
)
druggability_score: Mapped[Optional[float]] = mapped_column(Float, nullable=True)
volume: Mapped[Optional[float]] = mapped_column(Float, nullable=True) # in cubic angstroms
detection_method: Mapped[str] = mapped_column(
String(20), nullable=False, default="manual" # "fpocket", "manual", "ligand_based"
)
created_at: Mapped[datetime] = mapped_column(
DateTime(timezone=True), server_default=func.now()
)
# Relationships
protein_structure: Mapped[ProteinStructure] = relationship(back_populates="binding_sites")
def __repr__(self) -> str:
return f"<BindingSite(id={self.id}, name='{self.name}', method='{self.detection_method}')>"
chemlib/models/alignment.py¶
from __future__ import annotations
from datetime import datetime
from typing import Optional
from sqlalchemy import String, Text, Integer, Float, DateTime, ForeignKey, JSON
from sqlalchemy.orm import Mapped, mapped_column, relationship
from sqlalchemy.sql import func
from chemlib.models.base import Base
class SequenceAlignment(Base):
__tablename__ = "sequence_alignments"
id: Mapped[int] = mapped_column(primary_key=True)
name: Mapped[str] = mapped_column(String(300), nullable=False)
alignment_type: Mapped[str] = mapped_column(
String(20), nullable=False # "pairwise", "multiple"
)
method: Mapped[str] = mapped_column(
String(20), nullable=False # "biopython", "mafft", "clustalo"
)
input_sequences: Mapped[list] = mapped_column(
JSON, nullable=False
# [{"id": "P00533", "name": "EGFR_HUMAN", "sequence": "MRPSG..."}, ...]
)
alignment_data: Mapped[str] = mapped_column(
Text, nullable=False # Aligned FASTA format
)
score: Mapped[Optional[float]] = mapped_column(Float, nullable=True)
identity_pct: Mapped[Optional[float]] = mapped_column(Float, nullable=True)
num_sequences: Mapped[int] = mapped_column(Integer, nullable=False)
created_at: Mapped[datetime] = mapped_column(
DateTime(timezone=True), server_default=func.now()
)
def __repr__(self) -> str:
return f"<SequenceAlignment(id={self.id}, name='{self.name}', type='{self.alignment_type}')>"
class StructuralAlignment(Base):
__tablename__ = "structural_alignments"
id: Mapped[int] = mapped_column(primary_key=True)
name: Mapped[str] = mapped_column(String(300), nullable=False)
structure1_id: Mapped[int] = mapped_column(
ForeignKey("protein_structures.id", ondelete="CASCADE"), nullable=False
)
structure2_id: Mapped[int] = mapped_column(
ForeignKey("protein_structures.id", ondelete="CASCADE"), nullable=False
)
method: Mapped[str] = mapped_column(
String(20), nullable=False # "tmalign", "superimposer"
)
tm_score: Mapped[Optional[float]] = mapped_column(Float, nullable=True)
rmsd: Mapped[Optional[float]] = mapped_column(Float, nullable=True)
aligned_length: Mapped[Optional[int]] = mapped_column(Integer, nullable=True)
rotation_matrix: Mapped[Optional[list]] = mapped_column(
JSON, nullable=True # [[r11,r12,r13],[r21,r22,r23],[r31,r32,r33]]
)
translation_vector: Mapped[Optional[list]] = mapped_column(
JSON, nullable=True # [tx, ty, tz]
)
aligned_pdb_data: Mapped[Optional[str]] = mapped_column(
Text, nullable=True # Transformed structure2 PDB data for overlay
)
created_at: Mapped[datetime] = mapped_column(
DateTime(timezone=True), server_default=func.now()
)
# Relationships
structure1: Mapped[ProteinStructure] = relationship(
foreign_keys=[structure1_id]
)
structure2: Mapped[ProteinStructure] = relationship(
foreign_keys=[structure2_id]
)
def __repr__(self) -> str:
return f"<StructuralAlignment(id={self.id}, tm_score={self.tm_score})>"
Note: Import ProteinStructure from chemlib.models.protein at the module level or use string-based relationship references to avoid circular imports.
Pydantic Schemas¶
chemlib/schemas/protein.py¶
from __future__ import annotations
from datetime import datetime
from typing import Optional
from pydantic import BaseModel, ConfigDict, Field
# --- ProteinTarget ---
class ProteinTargetCreate(BaseModel):
"""Manual creation of a protein target."""
name: str
uniprot_id: str | None = None
gene_name: str | None = None
organism: str | None = None
description: str | None = None
sequence: str | None = None
ec_number: str | None = None
function_description: str | None = None
class ProteinTargetResponse(BaseModel):
id: int
uniprot_id: str | None
pdb_ids: list[str] | None
name: str
gene_name: str | None
organism: str | None
description: str | None
sequence: str | None
sequence_length: int | None
ec_number: str | None
function_description: str | None
created_at: datetime
updated_at: datetime
structure_count: int = 0 # Computed field
model_config = ConfigDict(from_attributes=True)
class ProteinTargetFilter(BaseModel):
name: str | None = None
gene_name: str | None = None
organism: str | None = None
ec_number: str | None = None
limit: int = 50
offset: int = 0
class ProteinTargetListResponse(BaseModel):
items: list[ProteinTargetResponse]
total: int
# --- ProteinStructure ---
class ProteinStructureCreate(BaseModel):
"""Upload a structure manually."""
protein_target_id: int
pdb_id: str | None = None
source: str = "user_upload" # "rcsb", "alphafold", "user_upload"
file_format: str = "pdb" # "pdb", "mmcif"
structure_data: str # Full PDB/mmCIF content
resolution: float | None = None
method: str | None = None # "xray", "nmr", "cryo_em", "predicted"
class ProteinStructureResponse(BaseModel):
id: int
protein_target_id: int
pdb_id: str | None
source: str
file_format: str
resolution: float | None
method: str | None
chains: list[str] | None
has_ligand: bool
ligand_ids: list[str] | None
created_at: datetime
# Note: structure_data excluded from list responses (too large)
model_config = ConfigDict(from_attributes=True)
class ProteinStructureDetailResponse(ProteinStructureResponse):
"""Includes full structure data for detail/download endpoints."""
structure_data: str
# --- BindingSite ---
class BindingSiteCreate(BaseModel):
"""Manually define a binding site."""
name: str
center_x: float
center_y: float
center_z: float
box_size_x: float = 20.0
box_size_y: float = 20.0
box_size_z: float = 20.0
residues: list[str] | None = None # ["A:GLU45", "A:ASP52"]
class BindingSiteFromLigand(BaseModel):
"""Define binding site from a co-crystallized ligand."""
ligand_id: str # e.g., "ATP"
padding: float = 5.0 # Angstroms around ligand
class BindingSiteResponse(BaseModel):
id: int
protein_structure_id: int
name: str
center_x: float
center_y: float
center_z: float
box_size_x: float
box_size_y: float
box_size_z: float
residues: list[str] | None
druggability_score: float | None
volume: float | None
detection_method: str
created_at: datetime
model_config = ConfigDict(from_attributes=True)
# --- Alignment ---
class SequenceAlignmentRequest(BaseModel):
"""Request a sequence alignment."""
name: str
sequences: list[SequenceInput]
method: str = "biopython" # "biopython" (pairwise), "mafft", "clustalo"
class SequenceInput(BaseModel):
id: str # identifier (UniProt ID, custom name)
name: str # display name
sequence: str # amino acid sequence
class SequenceAlignmentResponse(BaseModel):
id: int
name: str
alignment_type: str
method: str
num_sequences: int
score: float | None
identity_pct: float | None
alignment_data: str # aligned FASTA
created_at: datetime
model_config = ConfigDict(from_attributes=True)
class StructuralAlignmentRequest(BaseModel):
name: str
structure1_id: int
structure2_id: int
method: str = "tmalign" # "tmalign", "superimposer"
class StructuralAlignmentResponse(BaseModel):
id: int
name: str
structure1_id: int
structure2_id: int
method: str
tm_score: float | None
rmsd: float | None
aligned_length: int | None
aligned_pdb_data: str | None
created_at: datetime
model_config = ConfigDict(from_attributes=True)
Service Layer¶
chemlib/services/protein_target_service.py¶
class ProteinTargetService:
"""Business logic for protein target management."""
async def import_from_uniprot(
self, db: AsyncSession, accession: str
) -> ProteinTargetResponse:
"""
Fetch a protein from UniProt REST API and store it.
Steps:
1. Check if target with this uniprot_id already exists → return existing
2. Call UniProt REST API: GET https://rest.uniprot.org/uniprotkb/{accession}.json
3. Parse response: extract proteinDescription.recommendedName.fullName,
gene.geneName.value, organism.scientificName, sequence.value,
sequence.length, proteinDescription.ecNumbers, comments (FUNCTION)
4. Also fetch associated PDB IDs from cross-references
5. Create ProteinTarget record
6. Return response
"""
async def import_from_pdb(
self, db: AsyncSession, pdb_id: str
) -> ProteinTargetResponse:
"""
Import a protein target by first looking up its UniProt mapping from RCSB,
then importing from UniProt. Falls back to PDB metadata if no UniProt mapping.
Steps:
1. Call RCSB API to get UniProt mapping:
GET https://data.rcsb.org/rest/v1/core/entry/{pdb_id}
2. Extract UniProt accessions from polymer_entities
3. If found, call import_from_uniprot()
4. If not, create target from PDB metadata (entity name, organism, sequence)
5. Also fetch and store the structure (delegates to ProteinStructureService)
"""
async def search_uniprot(
self, query: str, limit: int = 25
) -> list[dict]:
"""
Search UniProt for proteins matching a query string.
Does NOT store results — returns search hits for user selection.
Call: GET https://rest.uniprot.org/uniprotkb/search?query={query}&size={limit}&format=json
Returns: list of {accession, name, gene, organism, sequence_length}
"""
async def get(self, db: AsyncSession, target_id: int) -> ProteinTargetResponse | None:
"""Get a protein target by ID, including structure count."""
async def list_targets(
self, db: AsyncSession, filters: ProteinTargetFilter
) -> ProteinTargetListResponse:
"""List protein targets with optional filters and pagination."""
async def delete(self, db: AsyncSession, target_id: int) -> bool:
"""Delete a protein target and all associated structures (cascade)."""
async def update(
self, db: AsyncSession, target_id: int, data: ProteinTargetCreate
) -> ProteinTargetResponse:
"""Update protein target metadata."""
chemlib/services/protein_structure_service.py¶
class ProteinStructureService:
"""Business logic for protein structure management."""
async def fetch_from_rcsb(
self, db: AsyncSession, pdb_id: str, protein_target_id: int
) -> ProteinStructureResponse:
"""
Fetch a PDB structure from RCSB and store it.
Steps:
1. Download PDB file: GET https://files.rcsb.org/download/{pdb_id}.pdb
2. Parse with Biopython Bio.PDB.PDBParser to extract:
- Chain IDs
- Resolution (from header)
- Method (X-RAY, NMR, etc.)
- Ligand HET codes (non-standard residues)
3. Store full PDB content in structure_data
4. Create ProteinStructure record
"""
async def fetch_from_alphafold(
self, db: AsyncSession, uniprot_id: str, protein_target_id: int
) -> ProteinStructureResponse:
"""
Fetch a predicted structure from AlphaFold DB.
Steps:
1. GET https://alphafold.ebi.ac.uk/api/prediction/{uniprot_id}
2. Parse response to get PDB URL (pdbUrl field)
3. Download PDB file from that URL
4. Store with source="alphafold", method="predicted"
"""
async def upload_structure(
self, db: AsyncSession, protein_target_id: int,
file_data: str, file_format: str = "pdb",
pdb_id: str | None = None
) -> ProteinStructureResponse:
"""
Store a user-uploaded PDB/mmCIF structure.
Steps:
1. Validate the structure data (attempt to parse with Biopython)
2. Extract chain IDs, ligands, method from header
3. Store with source="user_upload"
"""
async def get(self, db: AsyncSession, structure_id: int) -> ProteinStructureDetailResponse | None:
"""Get a structure by ID, including full structure_data."""
async def get_chains(self, db: AsyncSession, structure_id: int) -> list[dict]:
"""
Parse the structure and return chain info.
Returns: [{"chain_id": "A", "length": 320, "first_residue": 1, "last_residue": 320}, ...]
"""
async def extract_sequence(
self, db: AsyncSession, structure_id: int, chain_id: str
) -> str:
"""
Extract amino acid sequence from a specific chain using Biopython.
Returns: one-letter amino acid sequence string.
"""
async def list_for_target(
self, db: AsyncSession, protein_target_id: int
) -> list[ProteinStructureResponse]:
"""List all structures associated with a protein target."""
async def delete(self, db: AsyncSession, structure_id: int) -> bool:
"""Delete a structure and all associated binding sites (cascade)."""
chemlib/services/binding_site_service.py¶
class BindingSiteService:
"""Business logic for binding site detection and management."""
async def detect_pockets(
self, db: AsyncSession, structure_id: int,
min_druggability: float = 0.0
) -> list[BindingSiteResponse]:
"""
Run Fpocket on a protein structure and store detected pockets.
Steps:
1. Get structure from DB, write structure_data to temp file
2. Run: fpocket -f /tmp/{pdb_id}.pdb
3. Parse Fpocket output:
- Read {pdb_id}_out/{pdb_id}_info.txt for pocket scores
- Read {pdb_id}_out/pockets/ for pocket PDB files
- For each pocket: extract center (average of alpha sphere coords),
bounding box, residues, druggability score, volume
4. Filter by min_druggability
5. Create BindingSite records with detection_method="fpocket"
6. Clean up temp files
7. Return sorted by druggability_score descending
"""
async def define_from_ligand(
self, db: AsyncSession, structure_id: int,
ligand_id: str, padding: float = 5.0
) -> BindingSiteResponse:
"""
Define a binding site based on a co-crystallized ligand's position.
Steps:
1. Parse structure, find HETATM records matching ligand_id
2. Compute ligand center of mass (center_x/y/z)
3. Compute bounding box of ligand atoms + padding on each side
4. Identify protein residues within padding distance of any ligand atom
5. Create BindingSite with detection_method="ligand_based"
"""
async def define_manual(
self, db: AsyncSession, structure_id: int,
data: BindingSiteCreate
) -> BindingSiteResponse:
"""
Store a manually defined binding site.
User provides center and box dimensions directly.
"""
async def get(self, db: AsyncSession, site_id: int) -> BindingSiteResponse | None:
"""Get a binding site by ID."""
async def list_for_structure(
self, db: AsyncSession, structure_id: int
) -> list[BindingSiteResponse]:
"""List all binding sites for a structure."""
async def delete(self, db: AsyncSession, site_id: int) -> bool:
"""Delete a binding site."""
chemlib/services/alignment_service.py¶
class AlignmentService:
"""Sequence and structural alignment operations."""
async def pairwise_sequence_align(
self, db: AsyncSession,
name: str,
seq1: SequenceInput, seq2: SequenceInput,
method: str = "biopython",
matrix: str = "BLOSUM62",
gap_open: float = -10.0,
gap_extend: float = -0.5
) -> SequenceAlignmentResponse:
"""
Perform pairwise sequence alignment.
Method "biopython":
1. Use Bio.Align.PairwiseAligner with substitution_matrix=BLOSUM62
2. Set open_gap_score, extend_gap_score
3. Run aligner.align(seq1, seq2)
4. Take best alignment, compute identity percentage
5. Format as aligned FASTA
6. Store SequenceAlignment record with alignment_type="pairwise"
Method "mafft":
1. Write both sequences to temp FASTA file
2. Run: mafft --auto /tmp/input.fasta > /tmp/output.fasta
3. Parse output alignment
"""
async def multiple_sequence_align(
self, db: AsyncSession,
name: str,
sequences: list[SequenceInput],
method: str = "mafft"
) -> SequenceAlignmentResponse:
"""
Perform multiple sequence alignment.
Method "mafft" (recommended for 3+ sequences):
1. Write sequences to temp FASTA
2. Run: mafft --auto /tmp/input.fasta > /tmp/output.fasta
3. Parse aligned FASTA output
4. Compute pairwise identity percentages (average)
5. Store with alignment_type="multiple"
Method "clustalo":
1. Write sequences to temp FASTA
2. Run: clustalo -i /tmp/input.fasta -o /tmp/output.fasta --outfmt=fasta
3. Parse and store
"""
async def structural_align(
self, db: AsyncSession,
name: str,
structure1_id: int, structure2_id: int,
method: str = "tmalign",
chain1: str = "A", chain2: str = "A"
) -> StructuralAlignmentResponse:
"""
Perform structural alignment between two protein structures.
Method "tmalign" (via tmtools):
1. Get both structures from DB
2. Parse with Biopython, extract CA atoms for specified chains
3. Build coordinate arrays (N x 3)
4. Call tmtools.tm_align(coords1, coords2, seq1, seq2)
5. Extract: TM-score, RMSD, aligned_length, rotation_matrix, translation_vector
6. Apply transformation to structure2 PDB data to generate aligned_pdb_data
7. Store StructuralAlignment record
Method "superimposer" (via Biopython):
1. Parse structures, extract CA atoms
2. Use Bio.PDB.Superimposer to compute RMSD and rotation/translation
3. Apply to all atoms of structure2
4. Store results
"""
async def get_sequence_alignment(
self, db: AsyncSession, alignment_id: int
) -> SequenceAlignmentResponse | None:
"""Get a sequence alignment by ID."""
async def get_structural_alignment(
self, db: AsyncSession, alignment_id: int
) -> StructuralAlignmentResponse | None:
"""Get a structural alignment by ID."""
async def list_sequence_alignments(
self, db: AsyncSession, limit: int = 50, offset: int = 0
) -> list[SequenceAlignmentResponse]:
"""List all sequence alignments."""
async def generate_alignment_image(
self, alignment_id: int
) -> bytes:
"""
Generate a static alignment image (PNG) using pyMSAviz.
Reads alignment_data from the DB record, renders as a color-coded image.
Returns PNG bytes.
"""
Bioinformatics Utility Layer¶
chemlib/bioinformatics/pdb_parser.py¶
"""Pure PDB/mmCIF parsing utilities using Biopython. No DB access."""
from Bio.PDB import PDBParser, MMCIFParser, PDBIO
from Bio.PDB.Structure import Structure
from Bio.PDB.Chain import Chain
from Bio.PDB.Residue import Residue
import io
def parse_pdb_string(pdb_data: str, structure_id: str = "X") -> Structure:
"""Parse PDB format string into Biopython Structure object."""
parser = PDBParser(QUIET=True)
handle = io.StringIO(pdb_data)
return parser.get_structure(structure_id, handle)
def parse_mmcif_string(mmcif_data: str, structure_id: str = "X") -> Structure:
"""Parse mmCIF format string into Biopython Structure object."""
parser = MMCIFParser(QUIET=True)
handle = io.StringIO(mmcif_data)
return parser.get_structure(structure_id, handle)
def extract_chains(structure: Structure) -> list[dict]:
"""
Extract chain info from a Biopython Structure.
Returns: [{"chain_id": "A", "length": 320, "residues": [...], "sequence": "MRPS..."}]
"""
def extract_ligands(structure: Structure) -> list[dict]:
"""
Extract non-standard residues (HETATMs) that are ligands.
Excludes water (HOH/WAT) and common ions.
Returns: [{"het_id": "ATP", "chain": "A", "res_num": 501, "num_atoms": 31}]
"""
def extract_sequence_from_chain(chain: Chain) -> str:
"""Extract one-letter amino acid sequence from a chain."""
def get_resolution(structure: Structure) -> float | None:
"""Extract resolution from PDB header, if available."""
def get_method(structure: Structure) -> str | None:
"""Extract experimental method from PDB header."""
def compute_center_of_mass(residues: list[Residue]) -> tuple[float, float, float]:
"""Compute center of mass from a list of residues."""
def get_residues_near_point(
structure: Structure, center: tuple[float, float, float],
radius: float, chain_id: str | None = None
) -> list[str]:
"""
Find residues within radius of a point.
Returns: ["A:GLU45", "A:ASP52", ...]
"""
def structure_to_pdb_string(structure: Structure) -> str:
"""Convert a Biopython Structure back to PDB format string."""
output = io.StringIO()
io_obj = PDBIO()
io_obj.set_structure(structure)
io_obj.save(output)
return output.getvalue()
chemlib/bioinformatics/sequence_tools.py¶
"""Sequence alignment utilities. No DB access."""
from Bio.Align import PairwiseAligner, substitution_matrices
import subprocess
import tempfile
from pathlib import Path
def pairwise_align_biopython(
seq1: str, seq2: str,
matrix: str = "BLOSUM62",
gap_open: float = -10.0,
gap_extend: float = -0.5
) -> dict:
"""
Pairwise sequence alignment using Biopython.
Returns: {
"aligned_seq1": "MRPS-GTAGC...",
"aligned_seq2": "MRP--GTAGC...",
"score": 245.0,
"identity_pct": 78.5,
"alignment_fasta": ">seq1\nMRPS-GTAGC...\n>seq2\nMRP--GTAGC...\n"
}
"""
def multiple_align_mafft(
sequences: list[dict], # [{"id": "...", "name": "...", "sequence": "..."}]
mafft_binary: str = "mafft"
) -> dict:
"""
MSA using MAFFT subprocess.
Writes input FASTA to temp file, runs MAFFT, parses output.
Returns: {
"alignment_fasta": ">seq1\nMR-PS...\n>seq2\nMRPPS...\n",
"num_sequences": 5,
"alignment_length": 320
}
"""
def multiple_align_clustalo(
sequences: list[dict],
clustalo_binary: str = "clustalo"
) -> dict:
"""MSA using Clustal Omega subprocess. Same interface as MAFFT."""
def compute_identity(aligned_seq1: str, aligned_seq2: str) -> float:
"""Compute percent identity from two aligned sequences (with gaps)."""
def format_alignment_fasta(
sequences: list[dict], # [{"id": "...", "aligned_sequence": "..."}]
) -> str:
"""Format aligned sequences as FASTA string."""
chemlib/bioinformatics/structural_tools.py¶
"""Structural alignment utilities using tmtools and Biopython."""
import numpy as np
from Bio.PDB import Superimposer
from Bio.PDB.Structure import Structure
def tm_align(
structure1: Structure, structure2: Structure,
chain1: str = "A", chain2: str = "A"
) -> dict:
"""
Structural alignment using TM-align (via tmtools).
Steps:
1. Extract CA atom coordinates from specified chains
2. Extract sequences from chains
3. Call tmtools.tm_align(coords1, coords2, seq1, seq2)
4. Return results
Returns: {
"tm_score": 0.85,
"rmsd": 1.42,
"aligned_length": 280,
"rotation_matrix": [[r11,...], [r21,...], [r31,...]],
"translation_vector": [tx, ty, tz],
}
"""
def superimpose_biopython(
structure1: Structure, structure2: Structure,
chain1: str = "A", chain2: str = "A"
) -> dict:
"""
Superimpose structure2 onto structure1 using Biopython Superimposer.
Uses CA atoms for alignment.
Returns same dict format as tm_align (without tm_score).
"""
def apply_transformation(
structure: Structure,
rotation_matrix: list[list[float]],
translation_vector: list[float]
) -> Structure:
"""
Apply rotation + translation to all atoms of a structure.
Returns a new transformed Structure.
"""
def extract_ca_coords(structure: Structure, chain_id: str) -> np.ndarray:
"""Extract CA atom coordinates as Nx3 numpy array."""
chemlib/bioinformatics/pocket_detection.py¶
"""Fpocket integration for binding pocket detection."""
import subprocess
import tempfile
from pathlib import Path
def run_fpocket(
pdb_data: str,
fpocket_binary: str = "fpocket"
) -> list[dict]:
"""
Run Fpocket on a PDB structure and parse results.
Steps:
1. Write pdb_data to temp file: /tmp/fpocket_{uuid}.pdb
2. Run: {fpocket_binary} -f /tmp/fpocket_{uuid}.pdb
3. Parse output directory: /tmp/fpocket_{uuid}_out/
- pockets/pocket{n}_atm.pdb for pocket atom coordinates
- fpocket_{uuid}_info.txt for scores (druggability, volume, etc.)
4. For each pocket:
- Compute center from alpha sphere coordinates
- Compute bounding box
- Extract residue list
- Read druggability score and volume
5. Clean up temp files
6. Return sorted by druggability_score descending
Returns: [{
"pocket_number": 1,
"center": (x, y, z),
"box_size": (sx, sy, sz),
"residues": ["A:GLU45", "A:ASP52"],
"druggability_score": 0.82,
"volume": 456.7,
"num_alpha_spheres": 45
}, ...]
"""
def parse_fpocket_info(info_file: Path) -> list[dict]:
"""Parse Fpocket info.txt file to extract per-pocket metrics."""
def parse_pocket_pdb(pocket_pdb: Path) -> dict:
"""Parse a pocket PDB file to extract center, residues, atoms."""
chemlib/bioinformatics/protein_prep.py¶
"""Protein preparation using PDBFixer."""
from pdbfixer import PDBFixer
import io
def fix_protein(pdb_data: str) -> str:
"""
Prepare a protein structure for docking.
Steps using PDBFixer:
1. Load PDB data
2. Find missing residues → add them
3. Find missing atoms → add them
4. Remove heterogens (keep water optionally)
5. Add hydrogens at pH 7.0
6. Return fixed PDB string
Returns: fixed PDB data as string
"""
def remove_water(pdb_data: str) -> str:
"""Remove all water molecules from PDB data."""
def remove_heterogens(pdb_data: str, keep_water: bool = False) -> str:
"""Remove all HETATM records (optionally keep water)."""
def add_hydrogens(pdb_data: str, ph: float = 7.0) -> str:
"""Add hydrogens at specified pH using PDBFixer."""
chemlib/bioinformatics/external_apis.py¶
"""HTTP clients for external bioinformatics databases."""
import httpx
class UniProtClient:
"""Client for UniProt REST API (https://rest.uniprot.org)."""
BASE_URL = "https://rest.uniprot.org"
async def fetch_entry(self, accession: str) -> dict:
"""
GET /uniprotkb/{accession}.json
Returns parsed JSON with protein metadata.
"""
async def search(self, query: str, limit: int = 25) -> list[dict]:
"""
GET /uniprotkb/search?query={query}&size={limit}&format=json
Returns list of search results.
"""
def parse_entry(self, data: dict) -> dict:
"""
Parse UniProt JSON entry into our internal format:
{
"accession": "P00533",
"name": "Epidermal growth factor receptor",
"gene_name": "EGFR",
"organism": "Homo sapiens",
"sequence": "MRPSGTAGAALL...",
"sequence_length": 1210,
"ec_number": "2.7.10.1",
"function": "Receptor tyrosine kinase...",
"pdb_ids": ["1M17", "3W2S", ...]
}
"""
class RCSBClient:
"""Client for RCSB PDB REST API (https://data.rcsb.org)."""
BASE_URL = "https://data.rcsb.org"
FILES_URL = "https://files.rcsb.org"
async def fetch_entry_info(self, pdb_id: str) -> dict:
"""GET /rest/v1/core/entry/{pdb_id} — entry metadata."""
async def download_pdb(self, pdb_id: str) -> str:
"""GET https://files.rcsb.org/download/{pdb_id}.pdb — raw PDB file."""
async def download_mmcif(self, pdb_id: str) -> str:
"""GET https://files.rcsb.org/download/{pdb_id}.cif — raw mmCIF file."""
async def get_uniprot_mapping(self, pdb_id: str) -> list[str]:
"""Extract UniProt accessions from PDB entry metadata."""
class AlphaFoldClient:
"""Client for AlphaFold DB API (https://alphafold.ebi.ac.uk/api)."""
BASE_URL = "https://alphafold.ebi.ac.uk/api"
async def fetch_prediction(self, uniprot_id: str) -> dict:
"""GET /prediction/{uniprot_id} — prediction metadata including PDB URL."""
async def download_pdb(self, uniprot_id: str) -> str:
"""Download the predicted PDB file."""
API Endpoints¶
Protein Targets — chemlib/api/targets.py¶
| Method | Endpoint | Description | Request Body | Response |
|---|---|---|---|---|
GET |
/api/targets/ |
List all protein targets | Query: ProteinTargetFilter | ProteinTargetListResponse |
POST |
/api/targets/ |
Create a protein target manually | ProteinTargetCreate | ProteinTargetResponse |
GET |
/api/targets/{id} |
Get target details | — | ProteinTargetResponse |
PUT |
/api/targets/{id} |
Update target metadata | ProteinTargetCreate | ProteinTargetResponse |
DELETE |
/api/targets/{id} |
Delete target + structures | — | 204 |
GET |
/api/targets/{id}/structures |
List structures for target | — | list[ProteinStructureResponse] |
Protein Structures — chemlib/api/structures.py¶
| Method | Endpoint | Description | Request Body | Response |
|---|---|---|---|---|
GET |
/api/structures/{id} |
Get structure details (incl. data) | — | ProteinStructureDetailResponse |
POST |
/api/structures/ |
Upload a structure | ProteinStructureCreate | ProteinStructureResponse |
DELETE |
/api/structures/{id} |
Delete structure | — | 204 |
GET |
/api/structures/{id}/chains |
Get chain info | — | list[dict] |
GET |
/api/structures/{id}/sequence/{chain} |
Extract sequence for chain | — | {"sequence": "MRPS..."} |
GET |
/api/structures/{id}/binding-sites |
List binding sites | — | list[BindingSiteResponse] |
POST |
/api/structures/{id}/binding-sites |
Define a binding site manually | BindingSiteCreate | BindingSiteResponse |
POST |
/api/structures/{id}/binding-sites/from-ligand |
Define from ligand | BindingSiteFromLigand | BindingSiteResponse |
POST |
/api/structures/{id}/detect-pockets |
Run Fpocket | Query: min_druggability | list[BindingSiteResponse] |
External Fetch — chemlib/api/targets.py (or separate)¶
| Method | Endpoint | Description | Response |
|---|---|---|---|
POST |
/api/fetch/uniprot/{accession} |
Import target from UniProt | ProteinTargetResponse |
POST |
/api/fetch/rcsb/{pdb_id} |
Fetch structure from RCSB | ProteinStructureResponse |
POST |
/api/fetch/alphafold/{uniprot_id} |
Fetch from AlphaFold DB | ProteinStructureResponse |
GET |
/api/search/uniprot |
Search UniProt | Query: q (string) |
Alignments — chemlib/api/alignments.py¶
| Method | Endpoint | Description | Request Body | Response |
|---|---|---|---|---|
POST |
/api/alignments/sequence |
Run sequence alignment | SequenceAlignmentRequest | SequenceAlignmentResponse |
GET |
/api/alignments/sequence/{id} |
Get alignment result | — | SequenceAlignmentResponse |
GET |
/api/alignments/sequence/{id}/image |
Get alignment image (PNG) | — | image/png |
GET |
/api/alignments/sequence |
List sequence alignments | Query: limit, offset | list[SequenceAlignmentResponse] |
POST |
/api/alignments/structure |
Run structural alignment | StructuralAlignmentRequest | StructuralAlignmentResponse |
GET |
/api/alignments/structure/{id} |
Get structural alignment | — | StructuralAlignmentResponse |
GET |
/api/alignments/structure |
List structural alignments | Query: limit, offset | list[StructuralAlignmentResponse] |
Visualization¶
3D Protein Viewer (3Dmol.js)¶
The existing 3Dmol.js integration is extended for protein structures. The viewer is a reusable component in chemlib/static/js/protein_viewer.js.
Viewer Modes:
| Mode | 3Dmol.js Style | Use Case |
|---|---|---|
| Cartoon | setStyle({cartoon: {color: 'spectrum'}}) |
Default protein view |
| Surface | addSurface(...) with transparency |
Show molecular surface |
| Ball-and-stick | setStyle({stick: {}, sphere: {scale: 0.3}}) |
Ligand detail |
| Ribbon | setStyle({cartoon: {style: 'trace'}}) |
Simplified backbone |
Binding Site Visualization:
- Box overlay: addBox({center: {x, y, z}, dimensions: {w, h, d}, color: 'green', opacity: 0.3})
- Surface coloring: color residues in the binding site differently from the rest of the protein
- Residue highlighting: setStyle({resi: [45, 52, 721], chain: 'A'}, {stick: {color: 'yellow'}})
Protein Viewer JavaScript API (protein_viewer.js):
class ProteinViewer {
constructor(containerId) { /* Initialize 3Dmol.GLViewer */ }
async loadProtein(structureId) {
// Fetch PDB data from /api/structures/{id}
// viewer.addModel(data, "pdb")
// viewer.setStyle({}, {cartoon: {color: 'spectrum'}})
}
showBindingSite(siteData) {
// Draw box around binding site
// Highlight residues
}
showLigand(ligandId) {
// Show co-crystallized ligand in ball-and-stick
}
overlayStructure(pdbData, color) {
// Add second structure for superposition view
// viewer.addModel(pdbData, "pdb")
// viewer.setStyle({model: 1}, {cartoon: {color: color}})
}
setStyle(style) { /* Switch between cartoon, surface, stick */ }
zoomToSite(siteData) { /* Center view on binding site */ }
clear() { /* Remove all models */ }
}
Sequence Alignment Viewer (BioJS MSA Viewer)¶
For interactive, scrollable sequence alignment visualization in the browser. Loaded from CDN or bundled.
<!-- alignment_viewer.html -->
<div id="msa-viewer"></div>
<script>
// Fetch alignment data
const response = await fetch(`/api/alignments/sequence/${alignmentId}`);
const data = await response.json();
// Initialize MSA viewer
const msa = require("msa");
const viewer = msa({
el: document.getElementById("msa-viewer"),
seqs: parseFasta(data.alignment_data),
colorscheme: {"scheme": "clustal"}, // or "zappo", "hydrophobicity"
vis: {
conserv: true, // Show conservation track
overviewbox: true // Show minimap
}
});
viewer.render();
</script>
Color Schemes: Clustal (default), Zappo (physicochemical), Hydrophobicity, Taylor, Buried.
Static Alignment Images (pyMSAviz)¶
For server-side rendering (e.g., reports, downloads).
# In alignment_service.py
from pymsaviz import MsaViz
def generate_alignment_image(alignment_fasta: str) -> bytes:
mv = MsaViz(alignment_fasta, wrap_length=80, show_count=True)
mv.set_plot_params(color_scheme="Clustal")
buf = io.BytesIO()
mv.savefig(buf, format="png", dpi=150)
return buf.getvalue()
Structural Superposition Viewer¶
When viewing a structural alignment result, the 3D viewer loads both structures:
// In protein_viewer.js
async function showStructuralAlignment(alignmentId) {
const data = await fetch(`/api/alignments/structure/${alignmentId}`).then(r => r.json());
// Load structure 1 (reference) in blue
const struct1 = await fetch(`/api/structures/${data.structure1_id}`).then(r => r.json());
viewer.addModel(struct1.structure_data, "pdb");
viewer.setStyle({model: 0}, {cartoon: {color: '#3498db'}});
// Load structure 2 (aligned/transformed) in red
viewer.addModel(data.aligned_pdb_data, "pdb");
viewer.setStyle({model: 1}, {cartoon: {color: '#e74c3c'}});
// Show metrics overlay
showMetrics({tm_score: data.tm_score, rmsd: data.rmsd, aligned_length: data.aligned_length});
viewer.zoomTo();
viewer.render();
}
UI Pages¶
Protein Browser (protein_browser.html)¶
┌─────────────────────────────────────────────────────────────────┐
│ Protein Target Library [Import from UniProt] │
├─────────────────────────────────────────────────────────────────┤
│ Search: [________________] [Organism: ▾] [EC: ▾] [Search] │
├─────────────────────────────────────────────────────────────────┤
│ ┌─────────┬──────────┬───────────┬──────────┬────────┬──────┐ │
│ │ Name │ Gene │ Organism │ UniProt │ # Str │ Act │ │
│ ├─────────┼──────────┼───────────┼──────────┼────────┼──────┤ │
│ │ EGFR │ EGFR │ H.sapiens │ P00533 │ 12 │ View │ │
│ │ BRAF │ BRAF │ H.sapiens │ P15056 │ 8 │ View │ │
│ │ ... │ │ │ │ │ │ │
│ └─────────┴──────────┴───────────┴──────────┴────────┴──────┘ │
│ ◀ 1 2 3 ... ▶ │
└─────────────────────────────────────────────────────────────────┘
Protein Detail (protein_detail.html)¶
┌─────────────────────────────────────────────────────────────────┐
│ ◀ Back to Library │
│ │
│ EGFR — Epidermal Growth Factor Receptor │
│ Gene: EGFR | Organism: Homo sapiens | UniProt: P00533 │
│ EC: 2.7.10.1 | Length: 1210 aa │
│ │
│ Function: Receptor tyrosine kinase binding ligands... │
│ │
│ ┌──────────────────────────────────────────────────────────┐ │
│ │ 3D Structure Viewer (3Dmol.js) │ │
│ │ │ │
│ │ [Cartoon ribbon of selected structure] │ │
│ │ │ │
│ │ Style: [Cartoon ▾] Chain: [A ▾] Show binding sites: ☑ │ │
│ └──────────────────────────────────────────────────────────┘ │
│ │
│ Structures: │
│ ┌──────┬────────┬──────────┬──────┬───────────┬─────────────┐ │
│ │ PDB │ Source │ Method │ Res │ Chains │ Actions │ │
│ ├──────┼────────┼──────────┼──────┼───────────┼─────────────┤ │
│ │ 1M17 │ RCSB │ X-ray │ 2.6Å │ A │ View|Sites │ │
│ │ AF- │ AlphaF │ Predicted│ — │ A │ View|Sites │ │
│ └──────┴────────┴──────────┴──────┴───────────┴─────────────┘ │
│ [+ Fetch from RCSB] [+ Fetch from AlphaFold] [+ Upload] │
│ │
│ Binding Sites (for 1M17): │
│ ┌───────────────┬────────────────┬───────┬────────────┬──────┐ │
│ │ Name │ Method │ Score │ Volume │ Act │ │
│ ├───────────────┼────────────────┼───────┼────────────┼──────┤ │
│ │ Pocket 1 │ Fpocket │ 0.89 │ 567 A³ │ View │ │
│ │ ATP site │ Ligand-based │ — │ 423 A³ │ View │ │
│ └───────────────┴────────────────┴───────┴────────────┴──────┘ │
│ [Detect Pockets (Fpocket)] [Define from Ligand] [Define Manual]│
│ │
│ Sequence: │
│ MRPSGTAGAALLALLAALCPASRALEEKKVCQGTSNKLTQLGTFEDHFLSLQRMFNNCEVVL... │
│ [Copy] [Align with...] [BLAST search] │
└─────────────────────────────────────────────────────────────────┘
Alignment Viewer (alignment_viewer.html)¶
┌─────────────────────────────────────────────────────────────────┐
│ Sequence Alignment: EGFR_HUMAN vs ERBB2_HUMAN │
│ Method: BLOSUM62 | Identity: 42.3% | Score: 856.0 │
│ │
│ ┌──────────────────────────────────────────────────────────┐ │
│ │ [Interactive MSA Viewer — BioJS] │ │
│ │ Color scheme: [Clustal ▾] Wrap: [80 ▾] │ │
│ │ │ │
│ │ EGFR_HUMAN MRPSGTAGAALLALLAALCPASRALEEKKVCQGTSN----KL │ │
│ │ ERBB2_HUM MELAALCRWGLLLALLPPGA--ASTQVCTGTDMKLRL---PN │ │
│ │ ** * * ** * * * * │ │
│ │ [scrollable, with conservation bars below] │ │
│ └──────────────────────────────────────────────────────────┘ │
│ │
│ [Download FASTA] [Download Image (PNG)] │
└─────────────────────────────────────────────────────────────────┘
Alembic Migration¶
The migration adds all new tables in a single migration file:
# alembic/versions/xxxx_add_protein_target_tables.py
def upgrade():
op.create_table(
"protein_targets",
sa.Column("id", sa.Integer, primary_key=True),
sa.Column("uniprot_id", sa.String(20), unique=True, index=True, nullable=True),
sa.Column("pdb_ids", sa.JSON, nullable=True),
sa.Column("name", sa.String(500), nullable=False),
sa.Column("gene_name", sa.String(100), nullable=True),
sa.Column("organism", sa.String(200), nullable=True),
sa.Column("description", sa.Text, nullable=True),
sa.Column("sequence", sa.Text, nullable=True),
sa.Column("sequence_length", sa.Integer, nullable=True),
sa.Column("ec_number", sa.String(50), nullable=True),
sa.Column("function_description", sa.Text, nullable=True),
sa.Column("created_at", sa.DateTime(timezone=True), server_default=sa.func.now()),
sa.Column("updated_at", sa.DateTime(timezone=True), server_default=sa.func.now()),
)
op.create_table(
"protein_structures",
sa.Column("id", sa.Integer, primary_key=True),
sa.Column("protein_target_id", sa.Integer, sa.ForeignKey("protein_targets.id", ondelete="CASCADE"), nullable=False, index=True),
sa.Column("pdb_id", sa.String(10), nullable=True, index=True),
sa.Column("source", sa.String(20), nullable=False),
sa.Column("file_format", sa.String(10), nullable=False, default="pdb"),
sa.Column("structure_data", sa.Text, nullable=False),
sa.Column("resolution", sa.Float, nullable=True),
sa.Column("method", sa.String(20), nullable=True),
sa.Column("chains", sa.JSON, nullable=True),
sa.Column("has_ligand", sa.Boolean, default=False),
sa.Column("ligand_ids", sa.JSON, nullable=True),
sa.Column("created_at", sa.DateTime(timezone=True), server_default=sa.func.now()),
)
op.create_table(
"binding_sites",
sa.Column("id", sa.Integer, primary_key=True),
sa.Column("protein_structure_id", sa.Integer, sa.ForeignKey("protein_structures.id", ondelete="CASCADE"), nullable=False, index=True),
sa.Column("name", sa.String(200), nullable=False),
sa.Column("center_x", sa.Float, nullable=False),
sa.Column("center_y", sa.Float, nullable=False),
sa.Column("center_z", sa.Float, nullable=False),
sa.Column("box_size_x", sa.Float, nullable=False, default=20.0),
sa.Column("box_size_y", sa.Float, nullable=False, default=20.0),
sa.Column("box_size_z", sa.Float, nullable=False, default=20.0),
sa.Column("residues", sa.JSON, nullable=True),
sa.Column("druggability_score", sa.Float, nullable=True),
sa.Column("volume", sa.Float, nullable=True),
sa.Column("detection_method", sa.String(20), nullable=False, default="manual"),
sa.Column("created_at", sa.DateTime(timezone=True), server_default=sa.func.now()),
)
op.create_table(
"sequence_alignments",
sa.Column("id", sa.Integer, primary_key=True),
sa.Column("name", sa.String(300), nullable=False),
sa.Column("alignment_type", sa.String(20), nullable=False),
sa.Column("method", sa.String(20), nullable=False),
sa.Column("input_sequences", sa.JSON, nullable=False),
sa.Column("alignment_data", sa.Text, nullable=False),
sa.Column("score", sa.Float, nullable=True),
sa.Column("identity_pct", sa.Float, nullable=True),
sa.Column("num_sequences", sa.Integer, nullable=False),
sa.Column("created_at", sa.DateTime(timezone=True), server_default=sa.func.now()),
)
op.create_table(
"structural_alignments",
sa.Column("id", sa.Integer, primary_key=True),
sa.Column("name", sa.String(300), nullable=False),
sa.Column("structure1_id", sa.Integer, sa.ForeignKey("protein_structures.id", ondelete="CASCADE"), nullable=False),
sa.Column("structure2_id", sa.Integer, sa.ForeignKey("protein_structures.id", ondelete="CASCADE"), nullable=False),
sa.Column("method", sa.String(20), nullable=False),
sa.Column("tm_score", sa.Float, nullable=True),
sa.Column("rmsd", sa.Float, nullable=True),
sa.Column("aligned_length", sa.Integer, nullable=True),
sa.Column("rotation_matrix", sa.JSON, nullable=True),
sa.Column("translation_vector", sa.JSON, nullable=True),
sa.Column("aligned_pdb_data", sa.Text, nullable=True),
sa.Column("created_at", sa.DateTime(timezone=True), server_default=sa.func.now()),
)
def downgrade():
op.drop_table("structural_alignments")
op.drop_table("sequence_alignments")
op.drop_table("binding_sites")
op.drop_table("protein_structures")
op.drop_table("protein_targets")
Testing Strategy¶
| Component | Test Type | Fixtures |
|---|---|---|
| ORM models | Unit | In-memory SQLite |
| Bioinformatics utils | Unit | Sample PDB files, known sequences |
| External API clients | Unit (mocked) | Mocked httpx responses |
| Services | Integration | SQLite + mocked external APIs |
| API routes | E2E | Full stack with SQLite, mocked external APIs |
| Fpocket integration | Integration | Requires fpocket binary (skip if not installed) |
Sample Test Fixtures¶
# tests/conftest.py additions
SAMPLE_PDB_DATA = """HEADER HYDROLASE 01-JAN-00 XXXX
ATOM 1 N ALA A 1 1.000 2.000 3.000 1.00 0.00 N
ATOM 2 CA ALA A 1 2.000 3.000 4.000 1.00 0.00 C
...
END
"""
SAMPLE_SEQUENCE = "MRPSGTAGAALLALLAALCPASRALEEKKVCQGTSNKLTQLGTFEDHFLSLQ"
@pytest.fixture
async def protein_target(db_session):
target = ProteinTarget(
name="Test Protein",
uniprot_id="P00533",
gene_name="EGFR",
organism="Homo sapiens",
sequence=SAMPLE_SEQUENCE,
sequence_length=len(SAMPLE_SEQUENCE),
)
db_session.add(target)
await db_session.commit()
await db_session.refresh(target)
return target
@pytest.fixture
async def protein_structure(db_session, protein_target):
structure = ProteinStructure(
protein_target_id=protein_target.id,
pdb_id="1M17",
source="rcsb",
file_format="pdb",
structure_data=SAMPLE_PDB_DATA,
resolution=2.6,
method="xray",
chains=["A"],
has_ligand=True,
ligand_ids=["AQ4"],
)
db_session.add(structure)
await db_session.commit()
await db_session.refresh(structure)
return structure