Platform Overview — ChemLib Drug Discovery Platform¶
Vision¶
ChemLib began as a fragment-based drug design tool for curating chemical libraries, assembling molecules from fragments, and evaluating drug-likeness. The next evolution transforms ChemLib into a full Computer-Aided Drug Discovery (CADD) platform that covers the entire early-stage drug discovery workflow:
The platform retains ChemLib's existing strengths — compound management, BRICS assembly, scoring — and extends them with protein target management, structural biology tools, molecular docking, configurable screening pipelines, and an extensible plugin architecture. All modules share a single database, a unified API layer, and a cohesive web UI.
Click diagram to zoom and pan:
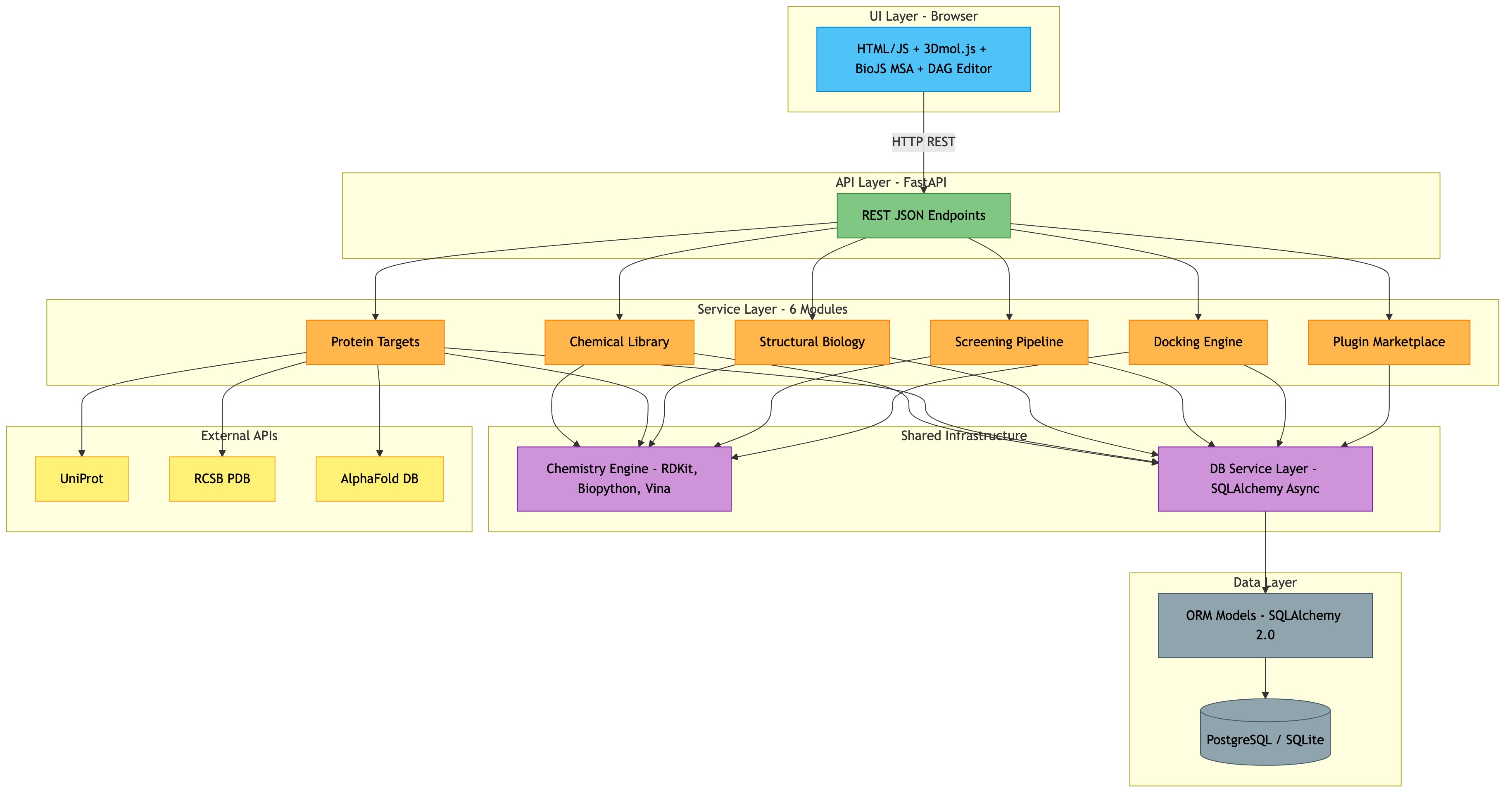
Platform Architecture (High-Level)¶
┌─────────────────────────────────────────────────────────────────────────────┐
│ WEB UI (Browser) │
│ HTML/JS + 3Dmol.js + BioJS MSA Viewer + DAG Pipeline Editor │
└──────────────────────────────────┬──────────────────────────────────────────┘
│ HTTP (REST JSON)
▼
┌─────────────────────────────────────────────────────────────────────────────┐
│ API LAYER (FastAPI) │
│ /api/compounds /api/fragments /api/assembly /api/scoring │
│ /api/targets /api/structures /api/alignments │
│ /api/docking /api/pipelines /api/plugins │
└──────────────────────────────────┬──────────────────────────────────────────┘
│
▼
┌─────────────────────────────────────────────────────────────────────────────┐
│ SERVICE LAYER │
│ │
│ ┌──────────────┐ ┌──────────────┐ ┌──────────────┐ ┌──────────────┐ │
│ │ Chemical │ │ Protein │ │ Structural │ │ Virtual │ │
│ │ Library │ │ Target │ │ Biology │ │ Screening │ │
│ │ Services │ │ Services │ │ Services │ │ Engine │ │
│ └──────────────┘ └──────────────┘ └──────────────┘ └──────────────┘ │
│ ┌──────────────┐ ┌──────────────┐ │
│ │ Docking & │ │ Plugin │ │
│ │ Scoring │ │ Marketplace │ │
│ │ Services │ │ Services │ │
│ └──────────────┘ └──────────────┘ │
└───────────┬──────────────────┬──────────────────────────────────────────────┘
│ │
▼ ▼
┌─────────────────────┐ ┌─────────────────────────────────────────────────┐
│ Chemistry Layer │ │ DB Service Layer (CRUD) │
│ RDKit, Biopython, │ │ SQLAlchemy 2.0 async │
│ Vina, Fpocket, │ │ │
│ PLIP, meeko │ │ CompoundDBService, FragmentDBService, │
│ │ │ ProteinTargetDBService, StructureDBService, │
│ │ │ PipelineDBService, DockingDBService, │
│ │ │ PluginDBService │
└─────────────────────┘ └──────────────────────┬──────────────────────────┘
│
▼
┌─────────────────────────────────────────────────┐
│ ORM MODELS (SQLAlchemy 2.0) │
│ Compound, Fragment, AssembledMolecule, │
│ ProteinTarget, ProteinStructure, BindingSite, │
│ SequenceAlignment, StructuralAlignment, │
│ Pipeline, PipelineRun, PipelineRunResult, │
│ DockingRun, DockingResult, │
│ FilterPluginRegistry │
└──────────────────────┬─────────────────────────┘
│
▼
┌─────────────────────────────────────────────────┐
│ DATABASE — PostgreSQL (prod) / SQLite (dev) │
└─────────────────────────────────────────────────┘
The Six Modules¶
1. Chemical Library (Existing — Phases 1-6)¶
The foundation. Manages compounds, fragments, BRICS decomposition, fragment assembly, conformer generation, energy minimization, drug-likeness scoring, and 3D visualization. Fully operational.
Key components: CompoundService, FragmentService, AssemblyService, ConformerService, ScoringService, VizService.
2. Protein Target Library (Phase 7)¶
Store, browse, and manage protein targets. Import from UniProt (sequence + metadata), RCSB PDB (structures), and AlphaFold DB (predicted structures). Each target can have multiple associated structures. Structures store the full PDB/mmCIF content for offline use.
Key components: ProteinTargetService, ProteinStructureService.
3. Structural Biology Tools (Phase 8)¶
Sequence alignment (pairwise via Biopython, multiple via MAFFT/Clustal Omega), structural alignment (TM-align via tmtools, Superimposer via Biopython), and homology search (BLAST, Foldseek). Visualize alignments interactively in the browser.
Key components: AlignmentService, HomologyService.
4. Virtual Screening Engine (Phase 11)¶
A configurable, DAG-based filtering pipeline that progressively narrows a compound library against a protein target. Users visually build pipelines by dragging filter nodes and connecting them. The executor processes compounds in batches, tracking pass/fail at each stage.
Key components: PipelineService, PipelineExecutor, FilterPlugin protocol.
5. Docking & Scoring (Phases 9-10)¶
Binding site detection (Fpocket), protein preparation (PDBFixer), ligand preparation (meeko), molecular docking (AutoDock Vina), interaction analysis (PLIP), and results visualization. Integrates into the screening pipeline as a docking filter node.
Key components: BindingSiteService, DockingService, InteractionService, ProteinPrepService.
6. Plugin Marketplace (Phase 12)¶
An extensible architecture for third-party tools. Plugins implement Protocol classes (FilterPlugin, DockingPlugin, ADMEPlugin, etc.), register in the database, and become available in the pipeline builder. Discovery via Python entry points.
Key components: PluginRegistryService, Protocol classes.
Data Flow — End-to-End Drug Discovery Workflow¶
Click diagram to zoom and pan:
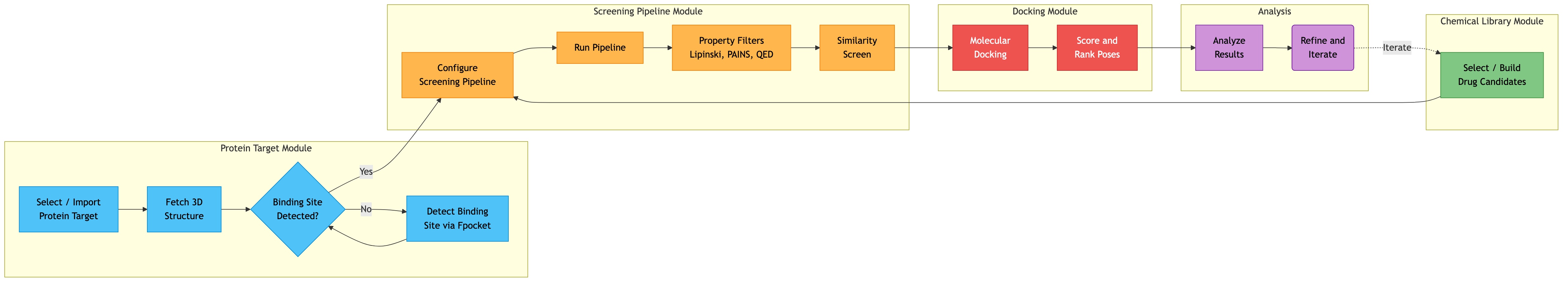
This shows how a user would use the platform to go from a protein target to docked lead compounds:
Step 1: SELECT TARGET
User imports a protein target from UniProt (e.g., EGFR, P00533)
→ ProteinTargetService.import_from_uniprot("P00533")
→ Stores: ProteinTarget record with sequence, metadata
│
▼
Step 2: FETCH STRUCTURE
User fetches a crystal structure from RCSB PDB (e.g., 1M17)
→ ProteinStructureService.fetch_from_rcsb("1M17")
→ Stores: ProteinStructure record with full PDB data
│
▼
Step 3: DETECT BINDING SITE
User runs Fpocket on the structure to find druggable pockets
→ BindingSiteService.detect_pockets(structure_id)
→ Stores: BindingSite records with center, box_size, druggability_score
User selects the most druggable pocket or defines one manually
│
▼
Step 4: PREPARE COMPOUND LIBRARY
User has already imported/assembled compounds (Chemical Library module)
OR: User assembles new candidates from the fragment library
→ CompoundService / AssemblyService
→ Available: hundreds to thousands of candidate compounds with SMILES
│
▼
Step 5: BUILD SCREENING PIPELINE
User opens the Pipeline Builder and creates a DAG:
[Library Input] → [Lipinski Filter] → [PAINS Filter] → [QED > 0.4]
→ [Tanimoto > 0.3 vs known EGFR inhibitor] → [Docking vs pocket]
→ [Interaction Filter (>= 2 H-bonds)] → [Results]
→ PipelineService.create_pipeline(definition)
│
▼
Step 6: RUN PIPELINE
User clicks "Run Pipeline" — selects the compound library as input
→ PipelineExecutor runs compounds through the DAG in batches
→ Each node filters: Lipinski (1000→800) → PAINS (800→750) → QED (750→400)
→ Similarity (400→150) → Docking (150→150, scored) → Interactions (150→50)
→ PipelineRun tracks progress, PipelineRunResult stores per-compound results
│
▼
Step 7: ANALYZE RESULTS
User views the funnel: 1000 → 800 → 750 → 400 → 150 → 50
User sorts the 50 surviving compounds by docking score
User clicks a compound to see:
- 3D pose overlaid on protein (3Dmol.js)
- Interaction diagram (H-bonds, hydrophobic contacts)
- Drug-likeness dashboard
- Assembly history (if assembled from fragments)
│
▼
Step 8: REFINE
User goes back to the fragment library, modifies promising leads
→ Swap a fragment, re-assemble, re-dock
→ Iterate until satisfied
Technology Stack¶
Existing (Chemical Library)¶
| Component | Technology |
|---|---|
| Language | Python 3.11+ |
| Database | PostgreSQL (prod) / SQLite (dev) |
| ORM | SQLAlchemy 2.0+ (async) |
| Migrations | Alembic |
| API | FastAPI + Pydantic v2 |
| Chemistry | RDKit |
| 3D Visualization | 3Dmol.js / py3Dmol |
| Frontend | HTML/JS + Jinja2 templates |
| Testing | pytest + httpx + pytest-asyncio |
New Additions¶
| Component | Technology | Purpose |
|---|---|---|
| Protein parsing | Biopython (Bio.PDB, Bio.SeqIO, Bio.Align) |
Parse PDB/mmCIF, sequence I/O, pairwise alignment |
| Structural alignment | tmtools | TM-align algorithm, TM-score computation |
| MSA (subprocess) | MAFFT (system binary) | Multiple sequence alignment, fast and accurate |
| MSA (subprocess, alt) | Clustal Omega (system binary) | Alternative MSA tool |
| Docking engine | vina (Python package) | AutoDock Vina molecular docking |
| Ligand preparation | meeko | SMILES/SDF → PDBQT for Vina |
| Receptor preparation | PDBFixer | Fix missing atoms/residues in PDB structures |
| Format conversion | openbabel-wheel | PDB ↔ PDBQT and other format conversions |
| Pocket detection | Fpocket (system binary) | Detect druggable binding pockets from protein structure |
| Interaction analysis | PLIP | Protein-Ligand Interaction Profiler (H-bonds, hydrophobic, pi-stacking) |
| 3D protein viewer | 3Dmol.js (already present) | Cartoon, surface, ball-and-stick rendering |
| Sequence alignment viz | BioJS MSA Viewer (JS) | Interactive, scrollable MSA visualization in browser |
| Static alignment images | pyMSAviz | Server-side alignment image generation (PNG/SVG) |
| ML ADME/Tox (optional) | DeepChem | ML-based ADME and toxicity predictions |
| Plugin system | Python Protocol classes + entry points | Extensible plugin architecture |
| External API client | httpx (already present) | Fetch from UniProt, RCSB, AlphaFold REST APIs |
System Binary Dependencies¶
These are not pip-installable and must be present on the system PATH:
| Binary | Install Method | Required For |
|---|---|---|
fpocket |
brew install fpocket / compile from source |
Binding site detection |
mafft |
brew install mafft / apt install mafft |
Multiple sequence alignment |
clustalo (optional) |
brew install clustal-omega |
Alternative MSA |
foldseek (optional) |
Download binary from Foldseek | Structural homology search |
Python Package Additions (requirements.txt)¶
biopython>=1.83
tmtools>=0.1.0
vina>=1.2.5
meeko>=0.5.0
openbabel-wheel>=3.1.1
pdbfixer>=1.9
plip>=2.3.0
pymsa>=0.7.0
pymsaviz>=0.4.0
deepchem>=2.7.0 # optional, for ML-based predictions
Shared Infrastructure¶
Database¶
All modules share a single PostgreSQL/SQLite database. New tables are added via Alembic migrations. The existing Base declarative base is extended with new models. Foreign key relationships link modules (e.g., DockingResult.compound_id → Compound.id).
API Layer¶
All new endpoints follow the same FastAPI patterns established in Phases 1-6. New routers are registered in chemlib/main.py. Each module gets its own router file under chemlib/api/.
Service Layer¶
New services follow the same stateless pattern. They call DB service methods for persistence and chemistry/bioinformatics utilities for computation. Services never access the database directly.
Configuration¶
New settings are added to chemlib/config.py:
class Settings(BaseSettings):
# ... existing settings ...
# Protein module
UNIPROT_API_BASE: str = "https://rest.uniprot.org"
RCSB_API_BASE: str = "https://data.rcsb.org"
ALPHAFOLD_API_BASE: str = "https://alphafold.ebi.ac.uk/api"
# Docking
VINA_EXHAUSTIVENESS: int = 32
VINA_NUM_POSES: int = 10
VINA_ENERGY_RANGE: float = 3.0
# Pipeline
PIPELINE_BATCH_SIZE: int = 100
PIPELINE_MAX_COMPOUNDS: int = 100_000
# Fpocket
FPOCKET_BINARY: str = "fpocket"
MAFFT_BINARY: str = "mafft"
Error Handling¶
New exception classes extending the existing hierarchy:
class ProteinNotFoundError(ChemLibError): ... # → 404
class StructureNotFoundError(ChemLibError): ... # → 404
class ExternalAPIError(ChemLibError): ... # → 502
class DockingError(ChemLibError): ... # → 500
class PipelineError(ChemLibError): ... # → 400
class PluginNotFoundError(ChemLibError): ... # → 404
class PluginConfigError(ChemLibError): ... # → 422
class BindingSiteError(ChemLibError): ... # → 400
Project Structure (Extended)¶
chemlib/
├── chemlib/
│ ├── models/
│ │ ├── compound.py # (existing)
│ │ ├── structure.py # (existing — Conformer)
│ │ ├── assembly.py # (existing)
│ │ ├── reaction.py # (existing)
│ │ ├── protein.py # NEW: ProteinTarget, ProteinStructure, BindingSite
│ │ ├── alignment.py # NEW: SequenceAlignment, StructuralAlignment
│ │ ├── pipeline.py # NEW: Pipeline, PipelineRun, PipelineRunResult
│ │ ├── docking.py # NEW: DockingRun, DockingResult
│ │ └── plugin.py # NEW: FilterPluginRegistry
│ ├── db/
│ │ └── service.py # Extended with new DB services
│ ├── api/
│ │ ├── compounds.py # (existing)
│ │ ├── fragments.py # (existing)
│ │ ├── assembly.py # (existing)
│ │ ├── visualization.py # (existing)
│ │ ├── scoring.py # (existing)
│ │ ├── targets.py # NEW: /api/targets/
│ │ ├── structures.py # NEW: /api/structures/
│ │ ├── alignments.py # NEW: /api/alignments/
│ │ ├── docking.py # NEW: /api/docking/
│ │ ├── pipelines.py # NEW: /api/pipelines/
│ │ └── plugins.py # NEW: /api/plugins/
│ ├── services/
│ │ ├── compound_service.py # (existing)
│ │ ├── fragment_service.py # (existing)
│ │ ├── assembly_service.py # (existing)
│ │ ├── conformer_service.py # (existing)
│ │ ├── scoring_service.py # (existing)
│ │ ├── viz_service.py # (existing)
│ │ ├── protein_target_service.py # NEW
│ │ ├── protein_structure_service.py # NEW
│ │ ├── binding_site_service.py # NEW
│ │ ├── protein_prep_service.py # NEW
│ │ ├── alignment_service.py # NEW
│ │ ├── docking_service.py # NEW
│ │ ├── interaction_service.py # NEW
│ │ ├── pipeline_service.py # NEW
│ │ ├── pipeline_executor.py # NEW
│ │ └── plugin_registry_service.py # NEW
│ ├── schemas/
│ │ ├── compound.py # (existing)
│ │ ├── fragment.py # (existing)
│ │ ├── assembly.py # (existing)
│ │ ├── scoring.py # (existing)
│ │ ├── protein.py # NEW
│ │ ├── alignment.py # NEW
│ │ ├── docking.py # NEW
│ │ ├── pipeline.py # NEW
│ │ └── plugin.py # NEW
│ ├── chemistry/ # (existing, unchanged)
│ ├── bioinformatics/ # NEW: Pure bioinformatics utilities
│ │ ├── __init__.py
│ │ ├── pdb_parser.py # PDB/mmCIF parsing via Biopython
│ │ ├── sequence_tools.py # Sequence alignment, format conversion
│ │ ├── structural_tools.py # TM-align, superimposition
│ │ ├── pocket_detection.py # Fpocket subprocess wrapper
│ │ ├── protein_prep.py # PDBFixer wrapper
│ │ └── external_apis.py # UniProt, RCSB, AlphaFold API clients
│ ├── docking/ # NEW: Docking utilities (no DB)
│ │ ├── __init__.py
│ │ ├── ligand_prep.py # SMILES → PDBQT via meeko
│ │ ├── receptor_prep.py # PDB → PDBQT
│ │ ├── vina_runner.py # AutoDock Vina Python API wrapper
│ │ └── interaction_analysis.py # PLIP wrapper
│ ├── plugins/ # NEW: Plugin system
│ │ ├── __init__.py
│ │ ├── protocols.py # Protocol classes (FilterPlugin, etc.)
│ │ ├── registry.py # Plugin discovery + registration
│ │ └── builtin/ # Built-in plugin implementations
│ │ ├── __init__.py
│ │ ├── property_filters.py
│ │ ├── similarity_filters.py
│ │ ├── adme_filters.py
│ │ ├── docking_filter.py
│ │ └── external_filters.py
│ ├── templates/
│ │ ├── base.html # (existing)
│ │ ├── index.html # Updated: platform dashboard
│ │ ├── protein_browser.html # NEW
│ │ ├── protein_detail.html # NEW
│ │ ├── alignment_viewer.html # NEW
│ │ ├── docking_viewer.html # NEW
│ │ ├── pipeline_builder.html # NEW
│ │ ├── pipeline_results.html # NEW
│ │ └── plugin_marketplace.html # NEW
│ └── static/
│ ├── js/
│ │ ├── viewer.js # (existing, extended)
│ │ ├── protein_viewer.js # NEW: protein-specific 3Dmol.js code
│ │ ├── pipeline_editor.js # NEW: DAG editor
│ │ └── msa_viewer.js # NEW: BioJS MSA integration
│ └── css/
│ └── platform.css # NEW: platform-wide styles
├── tests/
│ ├── test_protein/ # NEW
│ ├── test_alignment/ # NEW
│ ├── test_docking/ # NEW
│ ├── test_pipeline/ # NEW
│ └── test_plugins/ # NEW
└── scripts/
├── seed_fragments.py # (existing)
└── seed_plugins.py # NEW: Register built-in plugins
Cross-Module Integration Points¶
| From Module | To Module | Integration |
|---|---|---|
| Chemical Library | Screening Pipeline | Compounds/assembled molecules are pipeline input |
| Protein Target Library | Docking | Protein structures + binding sites are docking targets |
| Docking | Screening Pipeline | Docking is a filter node in the pipeline |
| Structural Biology | Protein Target Library | Alignments reference protein targets |
| Plugin Marketplace | Screening Pipeline | Plugins become pipeline filter nodes |
| All Modules | Visualization | 3Dmol.js for 3D, BioJS for sequences, charts for dashboards |
Naming Conventions¶
| Entity | Pattern | Example |
|---|---|---|
| ORM Models | PascalCase, singular |
ProteinTarget, DockingResult |
| DB Services | {Model}DBService |
ProteinTargetDBService |
| Business Services | {Domain}Service |
DockingService, AlignmentService |
| API Routers | chemlib/api/{plural_noun}.py |
targets.py, pipelines.py |
| Pydantic Schemas | {Model}Create, {Model}Response, {Model}Filter |
ProteinTargetCreate, DockingResultResponse |
| Bioinformatics utils | chemlib/bioinformatics/{function}.py |
pdb_parser.py, sequence_tools.py |
| Docking utils | chemlib/docking/{function}.py |
vina_runner.py, ligand_prep.py |
| Plugin implementations | chemlib/plugins/builtin/{category}.py |
property_filters.py |