Docking & Scoring Integration — Design Document¶
Overview¶
Molecular docking predicts how a small molecule (ligand) binds to a protein target. This module integrates open-source tools to provide a complete docking workflow within the ChemLib Platform:
- AutoDock Vina — primary docking engine (via Python API)
- meeko — ligand preparation (SMILES/RDKit Mol to PDBQT)
- PDBFixer — protein preparation (fix missing atoms, add hydrogens)
- Open Babel — format conversion (PDB to PDBQT for receptor)
- Fpocket — binding site detection (covered in Protein Target Library doc)
- PLIP — protein-ligand interaction profiling (H-bonds, hydrophobic, pi-stacking)
Docking Workflow¶
Click diagram to zoom and pan:
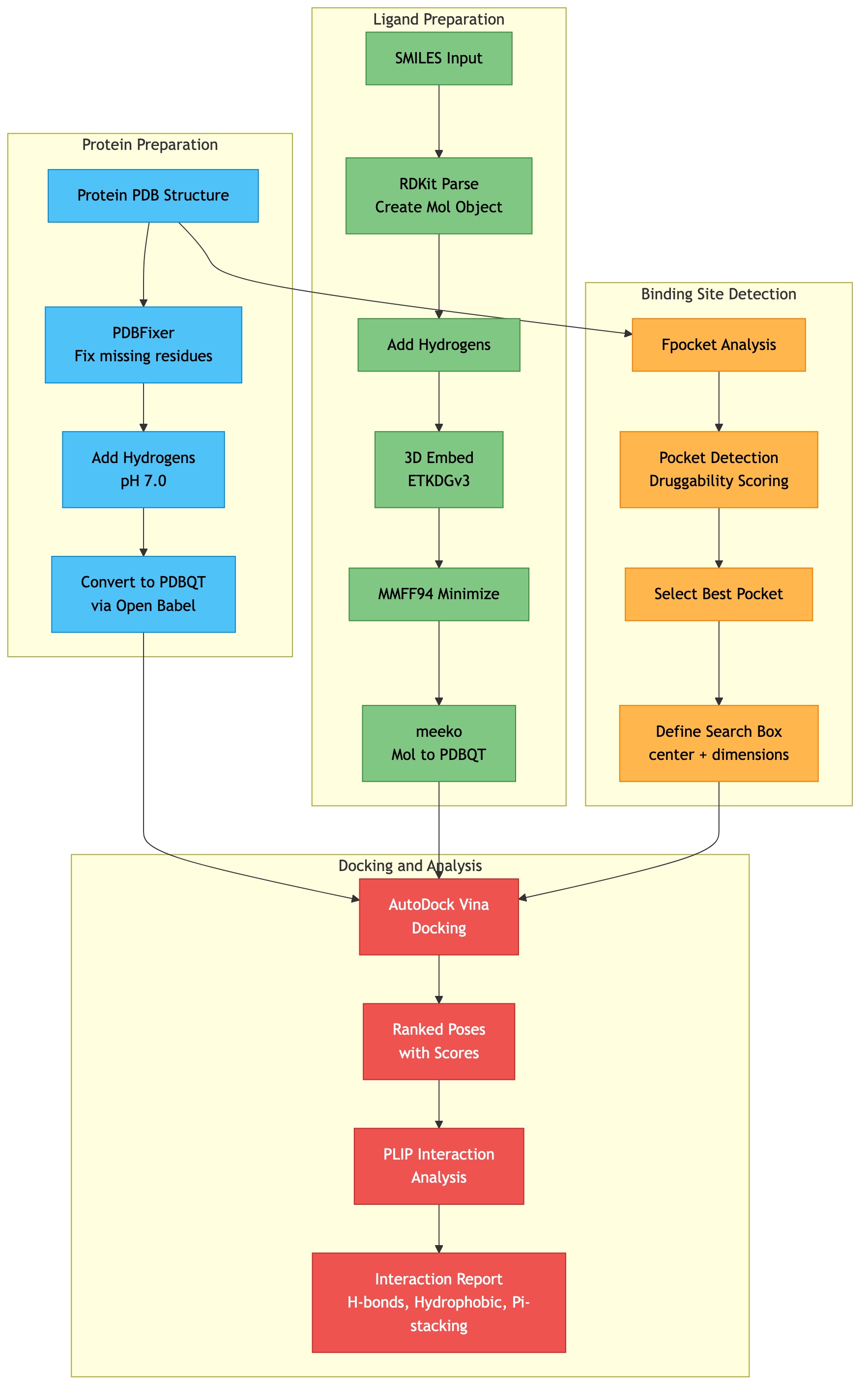
The complete docking workflow from raw inputs to analyzed results:
┌──────────────────────────────────────────────────────────────────────────┐
│ DOCKING WORKFLOW │
│ │
│ PROTEIN SIDE LIGAND SIDE │
│ ────────────── ──────────── │
│ │
│ PDB Structure SMILES / SDF │
│ │ │ │
│ ▼ ▼ │
│ ┌─────────────┐ ┌──────────────┐ │
│ │ PDBFixer │ │ RDKit Parse │ │
│ │ - Fix gaps │ │ - AddHs │ │
│ │ - Add H │ │ - 3D embed │ │
│ │ - Clean │ │ - MMFF94 min│ │
│ └──────┬──────┘ └──────┬───────┘ │
│ │ │ │
│ ▼ ▼ │
│ ┌─────────────┐ ┌──────────────┐ │
│ │ PDB → PDBQT │ │ Mol → PDBQT │ │
│ │ (Open Babel │ │ (meeko) │ │
│ │ or ADFR) │ │ │ │
│ └──────┬──────┘ └──────┬───────┘ │
│ │ │ │
│ ▼ ▼ │
│ ┌─────────────┐ ┌────────────┐ ┌──────────────┐ │
│ │ Receptor │ │ Binding │ │ Ligand │ │
│ │ PDBQT │──▶│ Site │◀────│ PDBQT │ │
│ └─────────────┘ │ (center, │ └──────────────┘ │
│ │ box_size)│ │
│ └─────┬──────┘ │
│ │ │
│ ▼ │
│ ┌────────────┐ │
│ │ AutoDock │ │
│ │ Vina │ │
│ │ - dock() │ │
│ └─────┬──────┘ │
│ │ │
│ ▼ │
│ ┌────────────┐ │
│ │ Ranked │ │
│ │ Poses │ │
│ │ (PDBQT) │ │
│ └─────┬──────┘ │
│ │ │
│ ▼ │
│ ┌────────────┐ │
│ │ PLIP │ │
│ │ Interaction│ │
│ │ Analysis │ │
│ └─────┬──────┘ │
│ │ │
│ ▼ │
│ ┌────────────┐ │
│ │ Results │ │
│ │ - Scores │ │
│ │ - Poses │ │
│ │ - Contacts │ │
│ └────────────┘ │
└──────────────────────────────────────────────────────────────────────────┘
Step-by-Step Detail¶
Step 1: Protein Preparation¶
# Input: Raw PDB structure data (from ProteinStructure.structure_data)
# Output: Cleaned PDB string, ready for conversion
# 1. Fix missing atoms and residues
from pdbfixer import PDBFixer
from openmm.app import PDBFile
import io
fixer = PDBFixer(pdbfile=io.StringIO(pdb_data))
fixer.findMissingResidues()
fixer.findMissingAtoms()
fixer.addMissingAtoms()
fixer.removeHeterogens(keepWater=False)
fixer.addHydrogens(pH=7.0)
output = io.StringIO()
PDBFile.writeFile(fixer.topology, fixer.positions, output)
fixed_pdb = output.getvalue()
Step 2: Receptor PDBQT Conversion¶
# Input: Fixed PDB string
# Output: PDBQT string (with partial charges and atom types)
# Option A: Open Babel (simpler)
import subprocess
# Write fixed PDB to temp file, convert with obabel
# obabel -ipdb receptor.pdb -opdbqt -O receptor.pdbqt -xr
# Option B: meeko/ADFR suite
# prepare_receptor -r receptor.pdb -o receptor.pdbqt
Step 3: Binding Site Definition¶
The binding site provides the search space for docking:
- center_x, center_y, center_z — center of the search box
- box_size_x, box_size_y, box_size_z — dimensions of the search box (Angstroms)
These come from the BindingSite table (detected via Fpocket, defined from ligand, or manual).
Step 4: Ligand Preparation¶
# Input: SMILES string
# Output: PDBQT string
from rdkit import Chem
from rdkit.Chem import AllChem
from meeko import MoleculePreparation, PDBQTWriterLegacy
# 1. Parse SMILES
mol = Chem.MolFromSmiles(smiles)
# 2. Add hydrogens
mol = Chem.AddHs(mol)
# 3. Generate 3D coordinates (ETKDGv3)
params = AllChem.ETKDGv3()
params.randomSeed = 42
AllChem.EmbedMolecule(mol, params)
# 4. Energy minimize (MMFF94)
AllChem.MMFFOptimizeMolecule(mol, maxIters=500)
# 5. Convert to PDBQT using meeko
preparator = MoleculePreparation()
mol_setup = preparator.prepare(mol)
pdbqt_string, is_ok, error_msg = PDBQTWriterLegacy.write_string(mol_setup)
Step 5: Docking with AutoDock Vina¶
# Input: Receptor PDBQT, Ligand PDBQT, Binding Site parameters
# Output: Ranked poses with scores
from vina import Vina
v = Vina(sf_name='vina')
# Set receptor
v.set_receptor(rigid_pdbqt=receptor_pdbqt_path)
# Set ligand
v.set_ligand_from_string(ligand_pdbqt_string)
# Set search space
v.compute_vina_maps(
center=[center_x, center_y, center_z],
box_size=[box_size_x, box_size_y, box_size_z]
)
# Dock
v.dock(
exhaustiveness=exhaustiveness, # Default: 32
n_poses=num_poses # Default: 10
)
# Get results
energies = v.energies() # Nx4 array: [score, lig_inter, lig_intra, torsions]
poses_pdbqt = v.poses(n_poses=num_poses) # PDBQT string with all poses
Step 6: Post-Processing and Interaction Analysis¶
# Input: Best docking pose (PDBQT) + protein structure
# Output: Interaction profile
# 1. Convert pose PDBQT back to PDB
# (via Open Babel or manual parsing)
# 2. Create complex PDB (protein + ligand)
complex_pdb = receptor_pdb + "\n" + ligand_pose_pdb
# 3. Run PLIP
from plip.structure.preparation import PDBComplex
complex = PDBComplex()
complex.load_pdb(complex_pdb_path)
complex.analyze()
for bsite_key, bsite in complex.interaction_sets.items():
interactions = {
"hydrogen_bonds": [
{
"donor": str(hb.d),
"acceptor": str(hb.a),
"distance": hb.distance_ad,
"angle": hb.angle,
"protein_residue": f"{hb.restype}{hb.resnr}",
}
for hb in bsite.hbonds_pdon + bsite.hbonds_ldon
],
"hydrophobic_contacts": [
{
"protein_residue": f"{hp.restype}{hp.resnr}",
"distance": hp.distance,
}
for hp in bsite.hydrophobic_contacts
],
"pi_stacking": [
{
"protein_residue": f"{ps.restype}{ps.resnr}",
"type": ps.type, # "P" (parallel) or "T" (T-shaped)
"distance": ps.distance,
}
for ps in bsite.pistacking
],
"salt_bridges": [
{
"protein_residue": f"{sb.restype}{sb.resnr}",
"distance": sb.distance,
"protein_is_positive": sb.protispos,
}
for sb in bsite.saltbridge_lneg + bsite.saltbridge_pneg
],
"water_bridges": [
{
"protein_residue": f"{wb.restype}{wb.resnr}",
"distance_protein_water": wb.distance_aw,
"distance_ligand_water": wb.distance_dw,
}
for wb in bsite.water_bridges
],
}
Service Layer¶
chemlib/services/docking_service.py¶
class DockingService:
"""
Orchestrates the full docking workflow:
protein preparation → ligand preparation → docking → results storage.
"""
async def prepare_receptor(
self, db: AsyncSession, structure_id: int,
remove_water: bool = True,
remove_heterogens: bool = True,
add_hydrogens: bool = True,
ph: float = 7.0
) -> dict:
"""
Prepare a protein structure for docking.
Steps:
1. Get ProteinStructure from DB
2. Fix with PDBFixer (missing atoms, hydrogens)
3. Convert to PDBQT (Open Babel subprocess)
4. Cache the prepared PDBQT (store in temp or in a new field)
Returns: {"receptor_pdbqt": "...", "fixed_pdb": "...", "num_atoms": 5000}
"""
async def prepare_ligand(
self, smiles: str
) -> dict:
"""
Prepare a small molecule for docking.
Steps:
1. Parse SMILES with RDKit
2. Add hydrogens
3. 3D embed (ETKDGv3)
4. MMFF94 minimize
5. Convert to PDBQT via meeko
Returns: {"ligand_pdbqt": "...", "num_atoms": 25, "num_rotatable": 4}
"""
async def dock(
self, db: AsyncSession,
structure_id: int,
binding_site_id: int,
ligand_smiles: list[str],
compound_ids: list[int | None] | None = None,
assembled_molecule_ids: list[int | None] | None = None,
exhaustiveness: int = 32,
num_poses: int = 10,
energy_range: float = 3.0
) -> int: # Returns DockingRun.id
"""
Run docking for a batch of ligands against a protein binding site.
Steps:
1. Prepare receptor (or use cached PDBQT)
2. Load binding site parameters (center, box_size)
3. Create DockingRun record with status="running"
4. For each ligand SMILES:
a. Prepare ligand PDBQT (meeko)
b. Run Vina docking
c. Parse results (scores, poses)
d. Run PLIP on best pose (optional, can be deferred)
e. Create DockingResult record
5. Update DockingRun status to "completed"
6. Return DockingRun.id
"""
async def dock_single(
self, receptor_pdbqt: str,
ligand_pdbqt: str,
center: tuple[float, float, float],
box_size: tuple[float, float, float],
exhaustiveness: int = 32,
num_poses: int = 10
) -> dict:
"""
Dock a single ligand. Low-level method used by dock() and pipeline filters.
Returns: {
"scores": [-8.5, -7.2, -6.8, ...], # kcal/mol per pose
"poses_pdbqt": "MODEL 1\n...", # All poses in PDBQT format
"best_score": -8.5,
"num_poses": 10
}
"""
async def get_run(self, db: AsyncSession, run_id: int) -> DockingRunResponse | None:
"""Get docking run status."""
async def get_results(
self, db: AsyncSession, run_id: int,
sort_by: str = "score",
sort_order: str = "asc",
limit: int = 50,
offset: int = 0
) -> list[DockingResultResponse]:
"""Get ranked docking results for a run."""
async def get_result_detail(
self, db: AsyncSession, result_id: int
) -> DockingResultDetailResponse:
"""Get a single docking result with full pose data."""
chemlib/services/interaction_service.py¶
class InteractionService:
"""Protein-ligand interaction analysis using PLIP."""
async def analyze_interactions(
self, protein_pdb: str, ligand_pdb: str
) -> dict:
"""
Analyze interactions between a protein and a docked ligand.
Steps:
1. Combine protein and ligand PDB into a complex
2. Write to temp file
3. Run PLIP analysis
4. Parse results into structured dict
Returns: {
"hydrogen_bonds": [
{"donor": "LIG:O3", "acceptor": "A:ASP855:OD1", "distance": 2.8,
"angle": 165.0, "is_strong": True}
],
"hydrophobic_contacts": [
{"protein_atom": "A:LEU718:CD1", "ligand_atom": "LIG:C12",
"distance": 3.6}
],
"pi_stacking": [...],
"salt_bridges": [...],
"water_bridges": [...],
"summary": {
"num_hbonds": 3,
"num_hydrophobic": 8,
"num_pi_stacking": 1,
"num_salt_bridges": 0,
"num_water_bridges": 1,
"total_interactions": 13
}
}
"""
async def analyze_docking_result(
self, db: AsyncSession, result_id: int
) -> dict:
"""
Analyze interactions for a stored docking result.
Fetches the protein structure and docking pose, runs PLIP,
and updates the DockingResult.interactions field.
"""
Docking Utility Layer¶
chemlib/docking/ligand_prep.py¶
"""Ligand preparation utilities for molecular docking."""
from rdkit import Chem
from rdkit.Chem import AllChem
from meeko import MoleculePreparation, PDBQTWriterLegacy
def smiles_to_pdbqt(smiles: str, seed: int = 42) -> str:
"""
Convert SMILES to PDBQT string for Vina docking.
Steps:
1. Parse SMILES → Mol
2. AddHs
3. Embed 3D (ETKDGv3)
4. MMFF94 minimize (500 iterations)
5. meeko MoleculePreparation → PDBQT
Returns: PDBQT string
Raises:
- ValueError if SMILES cannot be parsed
- RuntimeError if 3D embedding fails
- RuntimeError if meeko preparation fails
"""
mol = Chem.MolFromSmiles(smiles)
if mol is None:
raise ValueError(f"Cannot parse SMILES: {smiles}")
mol = Chem.AddHs(mol)
params = AllChem.ETKDGv3()
params.randomSeed = seed
status = AllChem.EmbedMolecule(mol, params)
if status == -1:
# Fallback: try without distance geometry
AllChem.EmbedMolecule(mol, AllChem.ETKDG())
AllChem.MMFFOptimizeMolecule(mol, maxIters=500)
preparator = MoleculePreparation()
mol_setup = preparator.prepare(mol)
pdbqt_string, is_ok, error_msg = PDBQTWriterLegacy.write_string(mol_setup)
if not is_ok:
raise RuntimeError(f"meeko preparation failed: {error_msg}")
return pdbqt_string
def mol_to_pdbqt(mol: Chem.Mol) -> str:
"""
Convert RDKit Mol (with 3D coords) to PDBQT string.
Assumes the mol already has hydrogens and 3D coordinates.
"""
preparator = MoleculePreparation()
mol_setup = preparator.prepare(mol)
pdbqt_string, is_ok, error_msg = PDBQTWriterLegacy.write_string(mol_setup)
if not is_ok:
raise RuntimeError(f"meeko preparation failed: {error_msg}")
return pdbqt_string
def pdbqt_to_pdb(pdbqt_string: str) -> str:
"""
Convert PDBQT to PDB format (strip charge and type columns).
Simple text manipulation — removes the last two columns of ATOM/HETATM lines.
"""
lines = []
for line in pdbqt_string.split('\n'):
if line.startswith(('ATOM', 'HETATM')):
lines.append(line[:66]) # PDB format is 66 chars for ATOM records
elif line.startswith(('MODEL', 'ENDMDL', 'END', 'REMARK', 'TER')):
lines.append(line)
return '\n'.join(lines)
def split_pdbqt_poses(pdbqt_string: str) -> list[str]:
"""
Split a multi-model PDBQT into individual pose strings.
Returns: list of PDBQT strings, one per pose.
"""
poses = []
current_pose = []
for line in pdbqt_string.split('\n'):
if line.startswith('MODEL'):
current_pose = [line]
elif line.startswith('ENDMDL'):
current_pose.append(line)
poses.append('\n'.join(current_pose))
current_pose = []
elif current_pose:
current_pose.append(line)
return poses
chemlib/docking/receptor_prep.py¶
"""Receptor preparation utilities for molecular docking."""
import subprocess
import tempfile
from pathlib import Path
def pdb_to_pdbqt(pdb_data: str, remove_water: bool = True) -> str:
"""
Convert PDB format to PDBQT format for Vina receptor.
Uses Open Babel subprocess:
obabel -ipdb input.pdb -opdbqt -O output.pdbqt -xr
The -xr flag tells Open Babel this is a rigid receptor (no flexible residues).
Returns: PDBQT string
"""
with tempfile.NamedTemporaryFile(suffix=".pdb", mode='w', delete=False) as f_in:
f_in.write(pdb_data)
input_path = Path(f_in.name)
output_path = input_path.with_suffix('.pdbqt')
try:
cmd = [
"obabel",
"-ipdb", str(input_path),
"-opdbqt",
"-O", str(output_path),
"-xr" # rigid receptor
]
result = subprocess.run(cmd, capture_output=True, text=True, timeout=60)
if result.returncode != 0:
raise RuntimeError(f"Open Babel failed: {result.stderr}")
return output_path.read_text()
finally:
input_path.unlink(missing_ok=True)
output_path.unlink(missing_ok=True)
def prepare_receptor_full(
pdb_data: str,
remove_water: bool = True,
remove_heterogens: bool = True,
add_hydrogens: bool = True,
ph: float = 7.0
) -> tuple[str, str]:
"""
Full receptor preparation pipeline:
1. PDBFixer: fix missing atoms, remove water/heterogens, add Hs
2. Convert to PDBQT
Returns: (fixed_pdb, receptor_pdbqt)
"""
from chemlib.bioinformatics.protein_prep import fix_protein
fixed_pdb = fix_protein(pdb_data)
receptor_pdbqt = pdb_to_pdbqt(fixed_pdb, remove_water=remove_water)
return fixed_pdb, receptor_pdbqt
chemlib/docking/vina_runner.py¶
"""AutoDock Vina docking engine wrapper."""
import tempfile
from pathlib import Path
from typing import Optional
from vina import Vina
def dock_ligand(
receptor_pdbqt: str,
ligand_pdbqt: str,
center: tuple[float, float, float],
box_size: tuple[float, float, float],
exhaustiveness: int = 32,
num_poses: int = 10,
energy_range: float = 3.0,
seed: int = 42
) -> dict:
"""
Dock a single ligand against a receptor using AutoDock Vina.
Args:
receptor_pdbqt: Receptor in PDBQT format
ligand_pdbqt: Ligand in PDBQT format
center: (x, y, z) center of search box
box_size: (sx, sy, sz) dimensions of search box in Angstroms
exhaustiveness: Search thoroughness (8-128, higher = more exhaustive)
num_poses: Maximum number of poses to return
energy_range: Maximum energy difference from best pose (kcal/mol)
seed: Random seed for reproducibility
Returns: {
"scores": [-8.5, -7.2, -6.8, ...], # kcal/mol, sorted best to worst
"poses_pdbqt": "MODEL 1\\n...", # All poses in multi-model PDBQT
"best_score": -8.5,
"energies": [ # Detailed energy breakdown
{"total": -8.5, "inter": -9.1, "intra": 0.6, "torsion": 0.0},
...
],
"num_poses_found": 10
}
"""
# Write receptor to temp file (Vina requires file path)
with tempfile.NamedTemporaryFile(suffix=".pdbqt", mode='w', delete=False) as f:
f.write(receptor_pdbqt)
receptor_path = f.name
try:
v = Vina(sf_name='vina', seed=seed)
v.set_receptor(rigid_pdbqt=receptor_path)
v.set_ligand_from_string(ligand_pdbqt)
v.compute_vina_maps(
center=list(center),
box_size=list(box_size)
)
v.dock(exhaustiveness=exhaustiveness, n_poses=num_poses)
energies_array = v.energies() # Nx4 numpy array
poses_pdbqt = v.poses(n_poses=num_poses)
scores = energies_array[:, 0].tolist()
energies = [
{
"total": float(row[0]),
"inter": float(row[1]),
"intra": float(row[2]),
"torsion": float(row[3]) if len(row) > 3 else 0.0,
}
for row in energies_array
]
return {
"scores": scores,
"poses_pdbqt": poses_pdbqt,
"best_score": scores[0] if scores else None,
"energies": energies,
"num_poses_found": len(scores),
}
finally:
Path(receptor_path).unlink(missing_ok=True)
def score_ligand(
receptor_pdbqt: str,
ligand_pdbqt: str,
center: tuple[float, float, float],
box_size: tuple[float, float, float]
) -> float:
"""
Score a ligand pose without docking (single-point scoring).
Useful for rescoring or evaluating a specific pose.
Returns: Vina score in kcal/mol
"""
with tempfile.NamedTemporaryFile(suffix=".pdbqt", mode='w', delete=False) as f:
f.write(receptor_pdbqt)
receptor_path = f.name
try:
v = Vina(sf_name='vina')
v.set_receptor(rigid_pdbqt=receptor_path)
v.set_ligand_from_string(ligand_pdbqt)
v.compute_vina_maps(center=list(center), box_size=list(box_size))
energy = v.score()
return float(energy[0])
finally:
Path(receptor_path).unlink(missing_ok=True)
chemlib/docking/interaction_analysis.py¶
"""Protein-ligand interaction analysis using PLIP."""
import tempfile
from pathlib import Path
from plip.structure.preparation import PDBComplex
def analyze_complex(complex_pdb: str) -> dict:
"""
Analyze protein-ligand interactions using PLIP.
Args:
complex_pdb: PDB format string containing both protein and ligand.
The ligand should be a HETATM residue.
Returns: {
"hydrogen_bonds": [{
"donor_type": "protein" | "ligand",
"donor": "A:ASP855:OD1",
"acceptor": "LIG:N1",
"distance": 2.82,
"angle": 162.5,
}, ...],
"hydrophobic_contacts": [{
"protein_residue": "A:LEU718",
"protein_atom": "CD1",
"ligand_atom": "C12",
"distance": 3.58,
}, ...],
"pi_stacking": [{
"protein_residue": "A:PHE723",
"type": "parallel", # or "t-shaped"
"distance": 4.12,
"angle": 15.3,
"offset": 1.2,
}, ...],
"salt_bridges": [{
"protein_residue": "A:LYS745",
"distance": 3.94,
"protein_is_positive": True,
}, ...],
"water_bridges": [{
"protein_residue": "A:THR790",
"water_id": "HOH:401",
"distance_protein_water": 2.75,
"distance_ligand_water": 2.88,
}, ...],
"halogen_bonds": [{
"protein_residue": "A:MET793",
"distance": 3.21,
"halogen": "Cl",
}, ...],
"metal_complexes": [...],
"summary": {
"num_hbonds": 3,
"num_hydrophobic": 8,
"num_pi_stacking": 1,
"num_salt_bridges": 0,
"num_water_bridges": 1,
"num_halogen_bonds": 0,
"total_interactions": 13,
}
}
"""
with tempfile.NamedTemporaryFile(suffix=".pdb", mode='w', delete=False) as f:
f.write(complex_pdb)
complex_path = f.name
try:
mol = PDBComplex()
mol.load_pdb(complex_path)
mol.analyze()
results = {
"hydrogen_bonds": [],
"hydrophobic_contacts": [],
"pi_stacking": [],
"salt_bridges": [],
"water_bridges": [],
"halogen_bonds": [],
"metal_complexes": [],
"summary": {},
}
for bsite_key, bsite in mol.interaction_sets.items():
# Parse H-bonds (protein-donor and ligand-donor)
for hb in bsite.hbonds_pdon:
results["hydrogen_bonds"].append({
"donor_type": "protein",
"donor": f"{hb.reschain}:{hb.restype}{hb.resnr}:{hb.d.type}",
"acceptor": f"LIG:{hb.a.type}",
"distance": round(hb.distance_ad, 2),
"angle": round(hb.angle, 1),
})
for hb in bsite.hbonds_ldon:
results["hydrogen_bonds"].append({
"donor_type": "ligand",
"donor": f"LIG:{hb.d.type}",
"acceptor": f"{hb.reschain}:{hb.restype}{hb.resnr}:{hb.a.type}",
"distance": round(hb.distance_ad, 2),
"angle": round(hb.angle, 1),
})
# Parse hydrophobic contacts
for hp in bsite.hydrophobic_contacts:
results["hydrophobic_contacts"].append({
"protein_residue": f"{hp.reschain}:{hp.restype}{hp.resnr}",
"distance": round(hp.distance, 2),
})
# Parse pi-stacking
for ps in bsite.pistacking:
results["pi_stacking"].append({
"protein_residue": f"{ps.reschain}:{ps.restype}{ps.resnr}",
"type": "parallel" if ps.type == "P" else "t-shaped",
"distance": round(ps.distance, 2),
"angle": round(ps.angle, 1),
"offset": round(ps.offset, 2),
})
# Parse salt bridges
for sb in bsite.saltbridge_lneg + bsite.saltbridge_pneg:
results["salt_bridges"].append({
"protein_residue": f"{sb.reschain}:{sb.restype}{sb.resnr}",
"distance": round(sb.distance, 2),
"protein_is_positive": sb.protispos,
})
# Parse water bridges
for wb in bsite.water_bridges:
results["water_bridges"].append({
"protein_residue": f"{wb.reschain}:{wb.restype}{wb.resnr}",
"distance_protein_water": round(wb.distance_aw, 2),
"distance_ligand_water": round(wb.distance_dw, 2),
})
# Summary
results["summary"] = {
"num_hbonds": len(results["hydrogen_bonds"]),
"num_hydrophobic": len(results["hydrophobic_contacts"]),
"num_pi_stacking": len(results["pi_stacking"]),
"num_salt_bridges": len(results["salt_bridges"]),
"num_water_bridges": len(results["water_bridges"]),
"num_halogen_bonds": len(results["halogen_bonds"]),
"total_interactions": sum(
len(results[k]) for k in [
"hydrogen_bonds", "hydrophobic_contacts", "pi_stacking",
"salt_bridges", "water_bridges", "halogen_bonds"
]
),
}
return results
finally:
Path(complex_path).unlink(missing_ok=True)
Database Schema¶
chemlib/models/docking.py¶
from __future__ import annotations
from datetime import datetime
from typing import Optional
from sqlalchemy import String, Text, Integer, Float, DateTime, ForeignKey, JSON
from sqlalchemy.orm import Mapped, mapped_column, relationship
from sqlalchemy.sql import func
from chemlib.models.base import Base
class DockingRun(Base):
__tablename__ = "docking_runs"
id: Mapped[int] = mapped_column(primary_key=True)
protein_structure_id: Mapped[int] = mapped_column(
ForeignKey("protein_structures.id", ondelete="CASCADE"), nullable=False, index=True
)
binding_site_id: Mapped[int] = mapped_column(
ForeignKey("binding_sites.id", ondelete="CASCADE"), nullable=False, index=True
)
status: Mapped[str] = mapped_column(
String(20), nullable=False, default="pending"
# "pending", "preparing", "running", "completed", "failed"
)
num_ligands: Mapped[int] = mapped_column(Integer, nullable=False, default=0)
completed_ligands: Mapped[int] = mapped_column(Integer, nullable=False, default=0)
exhaustiveness: Mapped[int] = mapped_column(Integer, nullable=False, default=32)
num_poses: Mapped[int] = mapped_column(Integer, nullable=False, default=10)
energy_range: Mapped[float] = mapped_column(Float, nullable=False, default=3.0)
receptor_pdbqt: Mapped[Optional[str]] = mapped_column(Text, nullable=True) # Cached
started_at: Mapped[Optional[datetime]] = mapped_column(DateTime(timezone=True), nullable=True)
completed_at: Mapped[Optional[datetime]] = mapped_column(DateTime(timezone=True), nullable=True)
error_message: Mapped[Optional[str]] = mapped_column(Text, nullable=True)
created_at: Mapped[datetime] = mapped_column(
DateTime(timezone=True), server_default=func.now()
)
# Relationships
results: Mapped[list[DockingResult]] = relationship(
back_populates="run", cascade="all, delete-orphan"
)
def __repr__(self) -> str:
return f"<DockingRun(id={self.id}, status='{self.status}', num_ligands={self.num_ligands})>"
class DockingResult(Base):
__tablename__ = "docking_results"
id: Mapped[int] = mapped_column(primary_key=True)
run_id: Mapped[int] = mapped_column(
ForeignKey("docking_runs.id", ondelete="CASCADE"), nullable=False, index=True
)
compound_id: Mapped[Optional[int]] = mapped_column(
ForeignKey("compounds.id", ondelete="SET NULL"), nullable=True, index=True
)
assembled_molecule_id: Mapped[Optional[int]] = mapped_column(
ForeignKey("assembled_molecules.id", ondelete="SET NULL"), nullable=True, index=True
)
smiles: Mapped[str] = mapped_column(String(2000), nullable=False)
score: Mapped[float] = mapped_column(Float, nullable=False) # kcal/mol (best pose)
pose_rank: Mapped[int] = mapped_column(Integer, nullable=False, default=1)
pose_pdbqt: Mapped[Optional[str]] = mapped_column(Text, nullable=True) # Best pose PDBQT
all_poses_pdbqt: Mapped[Optional[str]] = mapped_column(Text, nullable=True) # All poses
energies: Mapped[Optional[list]] = mapped_column(JSON, nullable=True) # Energy breakdown
interactions: Mapped[Optional[dict]] = mapped_column(JSON, nullable=True) # PLIP results
created_at: Mapped[datetime] = mapped_column(
DateTime(timezone=True), server_default=func.now()
)
# Relationships
run: Mapped[DockingRun] = relationship(back_populates="results")
def __repr__(self) -> str:
return f"<DockingResult(id={self.id}, smiles='{self.smiles[:30]}...', score={self.score})>"
Pydantic Schemas¶
chemlib/schemas/docking.py¶
from __future__ import annotations
from datetime import datetime
from typing import Optional, Any
from pydantic import BaseModel, ConfigDict, Field
class DockingRunCreate(BaseModel):
"""Request to start a docking run."""
structure_id: int
binding_site_id: int
compound_ids: list[int] | None = None
assembled_molecule_ids: list[int] | None = None
smiles_list: list[str] | None = None # Direct SMILES input
exhaustiveness: int = Field(default=32, ge=8, le=128)
num_poses: int = Field(default=10, ge=1, le=50)
energy_range: float = Field(default=3.0, ge=1.0, le=10.0)
analyze_interactions: bool = True # Run PLIP on best pose
class DockingRunResponse(BaseModel):
id: int
protein_structure_id: int
binding_site_id: int
status: str
num_ligands: int
completed_ligands: int
exhaustiveness: int
num_poses: int
started_at: datetime | None
completed_at: datetime | None
error_message: str | None
model_config = ConfigDict(from_attributes=True)
class DockingResultResponse(BaseModel):
id: int
run_id: int
compound_id: int | None
assembled_molecule_id: int | None
smiles: str
score: float
pose_rank: int
interactions_summary: dict | None = None # Just the summary counts
created_at: datetime
model_config = ConfigDict(from_attributes=True)
class DockingResultDetailResponse(DockingResultResponse):
"""Full detail including pose data and interactions."""
pose_pdbqt: str | None
all_poses_pdbqt: str | None
energies: list[dict] | None
interactions: dict | None
class InteractionAnalysisResponse(BaseModel):
hydrogen_bonds: list[dict]
hydrophobic_contacts: list[dict]
pi_stacking: list[dict]
salt_bridges: list[dict]
water_bridges: list[dict]
halogen_bonds: list[dict]
summary: dict
API Endpoints¶
Docking — chemlib/api/docking.py¶
| Method | Endpoint | Description | Request Body | Response |
|---|---|---|---|---|
POST |
/api/docking/prepare-receptor/{structure_id} |
Prepare protein for docking | Query: remove_water, add_hydrogens | {"status": "prepared", "num_atoms": 5000} |
POST |
/api/docking/run |
Submit a docking job | DockingRunCreate | DockingRunResponse |
GET |
/api/docking/runs/{run_id} |
Get docking run status | — | DockingRunResponse |
GET |
/api/docking/runs/{run_id}/results |
Get ranked results | Query: sort_by, limit, offset | list[DockingResultResponse] |
GET |
/api/docking/results/{result_id} |
Get single result detail | — | DockingResultDetailResponse |
GET |
/api/docking/results/{result_id}/interactions |
Get interaction analysis | — | InteractionAnalysisResponse |
GET |
/api/docking/results/{result_id}/pose |
Get pose PDB for viewer | Query: pose_rank (default 1) | {"pdb_data": "...", "score": -8.5} |
POST |
/api/docking/results/{result_id}/analyze |
Re-run PLIP analysis | — | InteractionAnalysisResponse |
Example API Flow¶
# 1. Prepare receptor
POST /api/docking/prepare-receptor/42
→ {"status": "prepared", "num_atoms": 5000}
# 2. Submit docking job
POST /api/docking/run
{
"structure_id": 42,
"binding_site_id": 7,
"compound_ids": [1, 2, 3, 4, 5],
"exhaustiveness": 32,
"num_poses": 10,
"analyze_interactions": true
}
→ {"id": 1, "status": "running", "num_ligands": 5, ...}
# 3. Poll for completion
GET /api/docking/runs/1
→ {"id": 1, "status": "running", "completed_ligands": 3, "num_ligands": 5, ...}
# 4. Get results
GET /api/docking/runs/1/results?sort_by=score&sort_order=asc
→ [
{"id": 1, "smiles": "CCO", "score": -8.5, "compound_id": 3, ...},
{"id": 2, "smiles": "CC(=O)O", "score": -7.2, "compound_id": 1, ...},
...
]
# 5. View pose in 3D
GET /api/docking/results/1/pose
→ {"pdb_data": "ATOM 1 N ...", "score": -8.5}
# 6. View interactions
GET /api/docking/results/1/interactions
→ {"hydrogen_bonds": [...], "hydrophobic_contacts": [...], "summary": {"num_hbonds": 3, ...}}
Visualization¶
Docking Results Viewer (docking_viewer.html)¶
┌─────────────────────────────────────────────────────────────────────────┐
│ Docking Results: Run #1 — EGFR (1M17) vs Pocket 1 Status: Done │
│ 5 ligands docked | Exhaustiveness: 32 | Poses: 10 │
├─────────────────────────────────────────────────────────────────────────┤
│ │
│ ┌──────────────────────────────────────────────────────────────────┐ │
│ │ │ │
│ │ 3D VIEWER (3Dmol.js) │ │
│ │ │ │
│ │ [Protein in cartoon (gray) + │ │
│ │ Docked ligand in sticks (green) + │ │
│ │ Binding site surface (transparent blue) + │ │
│ │ H-bond dashes (yellow) + │ │
│ │ Hydrophobic contacts (orange dots)] │ │
│ │ │ │
│ │ Pose: [1 ▾] of 10 Score: -8.5 kcal/mol │ │
│ │ Style: [Cartoon ▾] Surface: [☑ Binding Site] Labels: [☑] │ │
│ └──────────────────────────────────────────────────────────────────┘ │
│ │
│ ┌──────────────────────────────────────────────────────────────────┐ │
│ │ INTERACTION DIAGRAM (2D) │ │
│ │ │ │
│ │ ASP855 ──── H ────── O (ligand) 2.8 A │ │
│ │ MET793 ──── H ────── N (ligand) 3.1 A │ │
│ │ LEU718 ···· hydrophobic ···· C12 3.6 A │ │
│ │ PHE723 ═══ pi-stack ═══ ring 4.1 A │ │
│ │ │ │
│ │ Summary: 3 H-bonds | 8 hydrophobic | 1 pi-stack | 0 salt │ │
│ └──────────────────────────────────────────────────────────────────┘ │
│ │
│ RESULTS TABLE │
│ ┌────┬───────────────────┬──────────┬──────────┬──────────┬─────────┐ │
│ │ # │ Compound │ Score │ H-bonds │ Contacts │ Select │ │
│ ├────┼───────────────────┼──────────┼──────────┼──────────┼─────────┤ │
│ │ 1 │ CPD-003 (CCO...) │ -8.5 │ 3 │ 12 │ [View] │ │
│ │ 2 │ CPD-001 (CC...) │ -7.2 │ 2 │ 9 │ [View] │ │
│ │ 3 │ CPD-005 (c1c...) │ -6.8 │ 2 │ 7 │ [View] │ │
│ │ 4 │ CPD-002 (OC...) │ -5.4 │ 1 │ 5 │ [View] │ │
│ │ 5 │ CPD-004 (N#C...) │ -4.1 │ 0 │ 3 │ [View] │ │
│ └────┴───────────────────┴──────────┴──────────┴──────────┴─────────┘ │
└─────────────────────────────────────────────────────────────────────────┘
3Dmol.js Docking Viewer Implementation¶
// chemlib/static/js/protein_viewer.js — docking mode additions
class ProteinViewer {
// ... (base class from Protein Target Library doc)
async showDockingResult(resultId) {
this.clear();
// Fetch protein structure
const run = await this.fetchRunInfo(resultId);
const structure = await fetch(`/api/structures/${run.protein_structure_id}`).then(r => r.json());
// Load protein (cartoon, gray)
this.viewer.addModel(structure.structure_data, "pdb");
this.viewer.setStyle({model: 0}, {
cartoon: {color: '#cccccc', opacity: 0.8}
});
// Fetch docking pose
const pose = await fetch(`/api/docking/results/${resultId}/pose`).then(r => r.json());
// Load ligand pose (sticks, green)
this.viewer.addModel(pose.pdb_data, "pdb");
this.viewer.setStyle({model: 1}, {
stick: {colorscheme: 'greenCarbon', radius: 0.15},
sphere: {scale: 0.25, colorscheme: 'greenCarbon'}
});
// Show binding site surface
const site = await fetch(`/api/structures/${run.protein_structure_id}/binding-sites`).then(r => r.json());
if (site.length > 0) {
const bs = site.find(s => s.id === run.binding_site_id);
if (bs) {
// Highlight binding site residues
const residueSelector = bs.residues.map(r => {
const [chain, res] = r.split(':');
return {chain: chain, resi: parseInt(res.replace(/\D/g, ''))};
});
this.viewer.addSurface(
this.viewer.SurfaceType.VDW,
{opacity: 0.15, color: '#3498db'},
{or: residueSelector},
{model: 0}
);
}
}
// Show interactions
const interactions = await fetch(`/api/docking/results/${resultId}/interactions`).then(r => r.json());
this.showInteractionLines(interactions);
this.viewer.zoomTo({model: 1});
this.viewer.render();
}
showInteractionLines(interactions) {
// H-bonds as yellow dashed lines
for (const hb of interactions.hydrogen_bonds) {
// Would need atom coordinates — simplification here
// In practice, extract coords from PDB data
}
// Hydrophobic contacts as orange dotted lines
// Pi-stacking as green parallel lines
}
switchPose(resultId, poseRank) {
// Fetch specific pose: GET /api/docking/results/{id}/pose?pose_rank={rank}
// Replace model 1 with new pose
}
}
Score Heatmap (Multiple Ligands)¶
When viewing results for a docking run with many ligands, show a heatmap:
// Using simple HTML table with color-coded cells
function renderScoreHeatmap(results) {
// results: [{smiles, score, num_hbonds, num_contacts}, ...]
// Color scale: dark green (best score) → yellow → red (worst score)
// Score range: typically -12 to 0 kcal/mol
}
Alembic Migration¶
# alembic/versions/xxxx_add_docking_tables.py
def upgrade():
op.create_table(
"docking_runs",
sa.Column("id", sa.Integer, primary_key=True),
sa.Column("protein_structure_id", sa.Integer, sa.ForeignKey("protein_structures.id", ondelete="CASCADE"), nullable=False, index=True),
sa.Column("binding_site_id", sa.Integer, sa.ForeignKey("binding_sites.id", ondelete="CASCADE"), nullable=False, index=True),
sa.Column("status", sa.String(20), nullable=False, default="pending"),
sa.Column("num_ligands", sa.Integer, nullable=False, default=0),
sa.Column("completed_ligands", sa.Integer, nullable=False, default=0),
sa.Column("exhaustiveness", sa.Integer, nullable=False, default=32),
sa.Column("num_poses", sa.Integer, nullable=False, default=10),
sa.Column("energy_range", sa.Float, nullable=False, default=3.0),
sa.Column("receptor_pdbqt", sa.Text, nullable=True),
sa.Column("started_at", sa.DateTime(timezone=True), nullable=True),
sa.Column("completed_at", sa.DateTime(timezone=True), nullable=True),
sa.Column("error_message", sa.Text, nullable=True),
sa.Column("created_at", sa.DateTime(timezone=True), server_default=sa.func.now()),
)
op.create_table(
"docking_results",
sa.Column("id", sa.Integer, primary_key=True),
sa.Column("run_id", sa.Integer, sa.ForeignKey("docking_runs.id", ondelete="CASCADE"), nullable=False, index=True),
sa.Column("compound_id", sa.Integer, sa.ForeignKey("compounds.id", ondelete="SET NULL"), nullable=True, index=True),
sa.Column("assembled_molecule_id", sa.Integer, sa.ForeignKey("assembled_molecules.id", ondelete="SET NULL"), nullable=True, index=True),
sa.Column("smiles", sa.String(2000), nullable=False),
sa.Column("score", sa.Float, nullable=False),
sa.Column("pose_rank", sa.Integer, nullable=False, default=1),
sa.Column("pose_pdbqt", sa.Text, nullable=True),
sa.Column("all_poses_pdbqt", sa.Text, nullable=True),
sa.Column("energies", sa.JSON, nullable=True),
sa.Column("interactions", sa.JSON, nullable=True),
sa.Column("created_at", sa.DateTime(timezone=True), server_default=sa.func.now()),
)
def downgrade():
op.drop_table("docking_results")
op.drop_table("docking_runs")
Testing Strategy¶
| Component | Test Type | Setup |
|---|---|---|
ligand_prep.py |
Unit | Known SMILES, verify PDBQT output format |
receptor_prep.py |
Unit | Small test PDB, requires Open Babel binary |
vina_runner.py |
Integration | Requires vina package, small receptor + ligand |
interaction_analysis.py |
Integration | Known complex PDB, verify interaction counts |
DockingService |
Integration | Mocked Vina (fast), real DB |
| API endpoints | E2E | Full stack, mocked docking (fast) |
Test Fixtures¶
# Small test protein (just a few residues) for fast tests
TEST_RECEPTOR_PDB = """HEADER TEST
ATOM 1 N ALA A 1 1.000 2.000 3.000 1.00 0.00 N
ATOM 2 CA ALA A 1 2.000 3.000 4.000 1.00 0.00 C
...
END
"""
# Known docking result for regression testing
TEST_LIGAND_SMILES = "c1ccc(cc1)C(=O)O" # Benzoic acid
EXPECTED_SCORE_RANGE = (-8.0, -3.0) # kcal/mol
Mocking Vina for Fast Tests¶
class MockVinaRunner:
"""Mock Vina for unit tests that don't need real docking."""
def dock_ligand(self, receptor_pdbqt, ligand_pdbqt, center, box_size, **kwargs):
return {
"scores": [-7.5, -6.2, -5.8],
"poses_pdbqt": "MODEL 1\nATOM...\nENDMDL\n",
"best_score": -7.5,
"energies": [{"total": -7.5, "inter": -8.0, "intra": 0.5, "torsion": 0.0}],
"num_poses_found": 3,
}
Performance Considerations¶
| Operation | Time per Compound | Notes |
|---|---|---|
| Ligand preparation (meeko) | 50-200 ms | 3D embedding is the bottleneck |
| Receptor preparation | 5-30 s (one-time) | Cache the PDBQT |
| Vina docking (exhaust=8) | 5-30 s | Fast mode for screening |
| Vina docking (exhaust=32) | 30 s - 5 min | Default, good balance |
| Vina docking (exhaust=128) | 5-30 min | High accuracy, final validation |
| PLIP analysis | 1-5 s | Per complex |
Optimization strategies:
- Cache prepared receptor PDBQT in DockingRun.receptor_pdbqt
- Use lower exhaustiveness (8-16) for initial screening, higher (32-128) for final candidates
- Parallelize ligand preparation (CPU-bound but fast)
- Batch docking can run multiple Vina instances concurrently (IO-bound per instance)
- Defer PLIP analysis to on-demand (only when user views a result)